
- About Us
- Information
-

The Author ensures that the research has been conducted responsibly and ethically with adherence to all relevant regulations. read more..
- For Authors
- For Reviewer
- Manuscript Guidelines
- Membership
- Publication Ethics
-
- Journals
- Reprints
- e-Books
- Videos
- Policies
- Contact Us
 COVID-19
COVID-19
- Submissions



Full Text
Modern Approaches in Drug Designing
In-Silico Screening of Novel Antitoxic Agents from Talinum Paniculatum as Inhibitors of Phospholipase A2 And Metalloproteinase Receptor for Antivenom Activity
Bakare OS1*, Praise SA2*, Olubode SO1, Kolawole AT2, Olusanya MM2 and Ikechukwu CE3
1Department of Biochemistry, Faculty of Science, Adekunle Ajasin University, Nigeria
2Department of Biochemistry, College of Biosciences, Federal University of Agriculture, Nigeria
3Nigerian Institute of Medical Research, Nigeria
*Corresponding author:Bakare OS, Department of Biochemistry, Faculty of Science, Adekunle Ajasin University, Nigeria Praise SA, Department of Biochemistry, College of Biosciences, Federal University of Agriculture, Nigeria
Submission: September 12, 2025;Published: October 13, 2025

ISSN: 2576-9170 Volume4 Issue 5
Abstract
Snake, scorpion, and spider envenomation remains a significant global health challenge, with Phospholipase A2 (PLA2) and metalloproteinases recognised as key enzymatic mediators of venom toxicity. Inhibition of these enzymes is therefore a crucial strategy in antivenom development. Although synthetic inhibitors such as varespladib, varespladib-methyl, and darapladib have shown promise, their cytotoxicity underscores the need for safer and more effective alternatives. This study explored the bioactive compounds of Talinum paniculatum as potential antivenom agents. Molecular screening revealed that rutin, kaempferol, quercetin, and talinumoside I exhibited strong binding affinities to PLA2 and metalloproteinases, outperforming reference inhibitors including varespladib, varespladib-methyl, darapladib, marimastat, and ilomastat. Subsequent Absorption, Distribution, Metabolism, Excretion and Toxicity (ADMET) analyses identified quercetin and kaempferol as superior candidates, demonstrating favourable pharmacokinetic and safety profiles. These findings suggest that quercetin and kaempferol may serve as effective inhibitors of PLA2 and metalloproteinases, and thus hold promise as plant-derived antivenom leads. Further in vitro and in vivo investigations are warranted to validate their therapeutic potential.
Keywords:Rutin; Quercetin; Kaempferol; Varespladib; Envenomation; ADMET
Introduction
Yearly, there are about 1.5 million reported cases of scorpion envenoming and there is a potential for an increment in this number due to exponential improvement of urbanization [1]. There is a variation in the degree of envenomation effect on the human body and this is dependent on some parameters, but symptoms of it include hemorrhage, nausea, fever, dermatitis and hyperthermia which can degenerate into respiratory or heart failure, necrosis, hemorrhage, edema, inflammation, paralysis, coma, Hypotension and many more pathological outcomes [1,2]. The spread of venom is facilitated by enzymes such as phospholipases and metalloprotease through the disintegration of molecules within the matrix and the later brings about selective posttranslational processing of toxins caused by envenomation [3,4]. SvPLA2 facilitates venom spread through hydrolysis of phospholipids, disruption of cell membranes, activation of inflammatory pathways, degradation of extracellular matrix and induction of necrosis. The early occurrence of death and other adverse effects encountered after untreated snakebite is also attributed to the action of PLA2 due to its negative impact on homeostatic mechanisms which are crucial for the circulation of oxygen and coagulation regulation [5-8]. The mode of action of PLA2 involves the hydrolyzation of glycerophospholipids at the position of SN-2 of phospholipids which constitutes the release of oleic acid, lysophospholipids and arachidonic acid [9,10]. The effects of PLA2 include pre-synaptic neurotoxicity, coagulotoxicity, cardiotoxicity, and myotoxicity [11,12]. Metalloproteinase is an important enzyme in envenomation research because it constitutes about 30% of the total protein content contained in the venom of numerous vipers and other snakes [13]. Metalloproteinase has been studied to cause skin hemorrhage, apoptotic, fibrinolytic, fibrinogenolytic, activation of prothrombin and Factor X and also the aggregation of platelet [14] which has established it has a significant target for antivenom study. Phospholipases and metalloproteinase are mediators of inflammation triggered by envenomation and their concentration can be adequately regulated by the presence and binding of inhibitors. The inhibition of PLA2 activity by Varespladib establishes it as a drug candidate for envenomation [15]. Methylvarespladib and varespladib have shown the potency of inhibiting PLA2 obtained from six continents [16] and so are important treatment for envenomation.
Darapladib is also an effective potent inhibitor of PLA2 and this has facilitated its usage for clinical conditions and envenomation treatment [17]. Marimastat and Ilomastat are svMP inhibitors and are being investigated as treatments to venom-induced pathogenesis. Talinum paniculatum also known as “major gomes” and “erva gorda” is a weed predominant in Brazilian Cerrado and it has been put to use in folk medicine and for food [18]. It has also been used for cardiovascular disorder treatment [19], gastrointestinal problems, skin infections, and wound healing [18]. Its numerous clinical importance and its richness in essential phytocompounds such as tannins, triterpenes, steroids, saponins, and phytosterols [18] prompted the choice of this plant for this antivenom study. With the estimation of 15% of the whole animal biodiversity being poisonous and having toxins [20] and the limited supply of antivenins in so many areas, it is imperative to study a more effective and accessible antivenom with no side effect as compared to contemporary treatments which are still underevaluated and under-investigated. An in-silico study {which includes molecular docking, Molecular Mechanics Generalized Born Surface Area evaluation (MM/GBSA) and ADMET study} was done for the investigation of compounds (contained in Talinum paniculatum) with better inhibitory effects on PLA2 and metalloprotease and the pharmacokinetics of the lead compounds in comparison to the standard drugs (Varespladib, methyl-varespladib and darapladib).
Methodology
Ligands and protein target: For the identification and experimentation of the inhibitors of PLA2 and metalloproteinase which can ultimately serve as an essential antivenom constituent, the ligands (compounds of Talinum paniculatum) and standards utilized for this experiment were mined from PubChem in 2D SDF format. The crystal structures of the protein targets (PLA2 and Metalloproteinase) were obtained from protein data bank (http:// www.rscb.org/). Their PDB ID are 1TK4 and 1KUG respectively.
Ligand preparation: The bioactive compounds derived from Talinum paniculatum were prepared using the LigPrep tool in Schrödinger (Release 2017). In total, 40 compounds were identified, comprising flavonoids (rutin, quercetin, kaempferol), triterpenoids (ursolic acid, oleanolic acid), sterols (stigmasterol, β-sitosterol), and fatty acids (palmitic acid, stearic acid). In addition, reference inhibitors including varespladib, varespladib-methyl, darapladib, marimastat, and ilomastat were incorporated into the study for comparative analysis.
Protein preparation and receptor grid generation: The protein targets used were prepared with protein preparation wizard (2020) in Schrodinger suite. This was done to ensure restrain minimization and optimization of structures. Correction of essential aspects such as bond orders, charges, hydrogen consistency and water removal were also done. The Ca2+ ion was introduced to the active site to make it optimal for docking (particularly, as Varespladib and varespladib methyl coordinate the Ca2+ ion). Thereafter, the receptor grid was generated for the docking protocol using the prepared structure.
Molecular docking: The grid generated was utilized as the site for docking the bioactive compounds prepared as well as the standards (Varespladib, Methyl-varespladib and Darapladib) to the protein targets. The Standard Precision (SP) and a more rigid {Extra Precision (XP)} levels of docking were utilized for the obtainment of optimum results. Four compounds having the best pose and docking score were selected and subjected to further computational analyses such as binding energy and pharmacokinetic profiling for comparison with the standards. The interaction between the ligandprotein complexes formed were later visualized and analyzed.
Molecular Mechanics / Generalized Born Surface Area (MM/GBSA): The calculation for binding free energy in ligandreceptor (L-R) interactions can be expressed as:
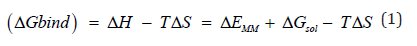
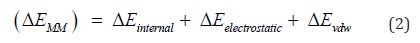
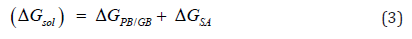
Upon binding, ΔEMM = the MM energy
ΔGsol = the solvation free energy
-TΔS = the conformational entropy
These are representations upon changes of the gas phase.
Furthermore, ΔEMM consists of ΔEinternal (including bond, angle, and dihedral energies), ΔEelectrostatic, and ΔEvdw (van der Waals) energies [21]. The summation of electrostatic solvation energy, non-electrostatic solvation component, ΔGPB/GB, and ΔGSA brings about ΔGsolv [21].
Frontal molecular orbital analysis: Frontier Molecular Orbital (FMO) theory is a concept in chemistry that helps explain and predict chemical reactivity based on the interaction between the Highest Occupied Molecular Orbital (HOMO) and the Lowest Unoccupied Molecular Orbital (LUMO) of reacting molecules. The HOMO and LUMO, being the most likely orbitals to be involved in chemical reactions, are crucial in estimating various chemical reactivity descriptors, such as Energy gap (Eg), Ionization potential(I), Electron Affinity (A), electronegativity (χ), electrophilicity (δ) global hardness (η), softness (S), and dipole moment (ω). These descriptors are calculated using the following equations:
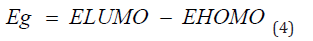
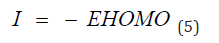
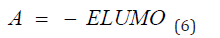
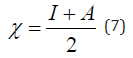
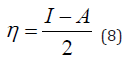
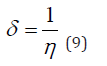
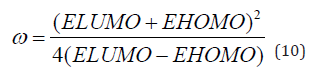
Pharmacokinetic profiling: The study of Absorption, distribution, metabolism, excretion, and toxicity of potential drug candidates is very essential for drug discovery and experimentation. SwissAdme (http://www.swissadme.ch) and Pro-tox II (http:// tox.charite.de/protox_II) were utilized for such study. Descriptors such as, bioavailability score, ability to inhibit cytochrome P450 enzymes, P-glycoprotein substrate candidacy, Topological Surface Area (TSA), blood brain barrier permeability, molecular weight of compounds, water solubility, lipophilicity, and absorption rate via the gastrointestinal tract were analyzed.
Results and discussion: Docking entails having an output of scoring functions obtained through algorithms that calculate the level of binding affinity involved in protein–ligand interaction and this computation subsequently generates a scoring function for the ranking of the most energetical protein-bound conformation in line with the individual ligands [22]. About 40 compounds were obtained from Talinum paniculatum as well as five standards (Varespladib, Varespladib methyl, Darapladib, Marimastat and Ilomastat) were screened against two protein targets (Phospholipase A2 and Metalloproteinase) to investigate their inhibitory effect via the binding affinities. The four lead compounds (rutin, kaempferol, quercetin and talinumoside1) showed a good inhibitory effect in comparison to the standards. Quercetin had the highest binding affinity to PLA2 with docking score of -9.033kcal/mol and Rutin was ranked highest for metalloproteinase with -12.953kcal/mol. The three other lead compounds showed very impressive inhibitory potential to the targets having docking scores in the range of -5.768 and -8.622kcal/mol for PLA2 (which were higher than the standards) and -6.974 and -9.145kcal/mol for metalloproteinase as illustrated in Figure 1. After thorough investigation of the ligands’ interaction with the targets, it was seen that with PLA2, rutin and quercetin interact with Phe5 through pi-pi stacking and hydrogen bond (H-bond) respectively. Kaempferol and quercetin interact with Hie48 via pi-pi stacking and H-bond. Rutin binds to Ser23 and Gly30 via hydrogen bonding. While the binding of kaempferol to Ash49 was also through H-bond. The interaction between Quercetin and the target also involves pi-pi stacking with Ash49 and Leu2. Arg72 of PLA2 forms an H-bond and salt bridge with talinumoside1 while Leu3 and Lys69 interacts with the ligand via H-bond. These interactions are shown in Figure 2.
Figure 1:Diagrammatic Representation of Molecular Docking Score (kcal/mol) of Screened ligands from Talinum paniculatum and standards to PLA 2 and Metalloproteinase Respectively.

Figure 2:2D Interactions of the Lead Compounds with Amino Acid Residues at the Active Sites of PLA 2 (a) Rutin, (b) Kaempferol, (c) Quercetin (d) Talinumoside1 (e) Varespladib (f) Varespladib Methyl (g) Darapladib.

The stability of rutin with metalloproteinase can be said to be due to the H-bond it forms with Glh145, Ala113 and Ala169 amongst other bonding. It is worth noting that Thr109 forms a hydrogen bond with rutin, kaempferol and talinumoside1 while Trp112 binds via pi-pi stacking to rutin and H-bond to it and talinumoside1. A pi-cation interaction occurred at the active pocket of metalloproteinase at Arg107 with rutin and the said amino acid also contacted talinumoside1 via H-bond. Asn108 established a hydrogen bond contact with Kaempferol and talinumoside1 while the later also formed a H-bond with Asp72. A pi-pi interaction of quercetin and kaempferol with metalloproteinase was established at His144. Quercetin also contacted the target via pi-cation and H-bond at Ser168 and Hip154. These interactions with metalloproteinase are illustrated in Figure 3.
Figure 3:2D Interactions of the Lead Compounds with Amino Acid Residues at the Active Sites of Metalloproteinase (a) Rutin, (b) Kaempferol, (c) Quercetin (d) Talinumoside I (e)Marimastat (f)Ilomastat.

Salt bridges play a fundamental role for the stability of secondary-structural elements [23]. The connection of several subunits of proteins is also a function that salt bridges perform in interactions [24]. The H-bond is very essential in protein-ligand interactions because of its support in ligand binding affinity using the mechanism of displacement of protein-bound water molecules and also has a role in protein folding and catalysis [25]. Pi-stackings interact via non-covalent force for stabilization purpose. Protein aromatic residues are important for the substrate binding and also stability when interacting via pi-stacking [26]. The manner at which the interacting molecules orientate (geometrically) determines the strength of aromatic stacking interactions [27]. For molecular interactions, pi-cation also utilizes noncovalent forces [28] and it forms a bond between monopoles (cations) and a quadrupole (π system) while also promoting protein structure. Furthermore, the binding energy of each ligand to the protein targets were investigated to know the level of spontaneity of each compound in complex with the receptor. It is worth noting that the more negative the value of the binding energy (ΔGbind) of a compound to a target is, the higher its spontaneity. With a binding energy of -52.145kcal/ mol, rutin can be said to be the most spontaneous compound to PLA2 with respect to this study. Kaempferol and quercetin (-50.132 and -48.907kcal/mol) had higher ΔGbind than the standards used, while talinumoside1 had a value of -41.995kcal/mol which is an impressive value in comparison with the standard as shown grammatically in Figure 4a. A closely related trend was observed with the ΔGbind of the ligands’ interaction with metalloproteinase. Although, Marimastat had the highest biding energy of -58.557 kcal/mol but it was closely followed by Rutin, quercetin and kaempferol which were ranked -57.712, -55.209 and -54.845kcal/ mol respectively while talinumoside1 having a value of -34.162 kcal/mol showed a fair level of spontaneity with respect to the standards. Molecular Mechanics/ Generalized Born Surface Area (MM/GBSA) method utilizes both molecular mechanics calculations and the continuum solvation models [21]. The scores of the MM/ GBSA protocol with metalloproteinase are shown in Figure 4b.
Figure 4:Diagrammatic Representation of MM-GBSA of Screened Ligands from Talinum paniculatum and Standard to PLA 2 and Metalloproteinase Respectively.

Frontier molecular orbital: Many studies have proven that energy quantization in molecular orbitals is a key component in predicting spontaneity and stability of chemical reactions [29,30]. The spatial distribution of electron in a molecule when two or more nuclei interact is known as molecular orbitals. During molecular interactions, electrons are distributed into orbitals according to specific energy level. The Highest Occupied Molecular Orbitals (HOMO) signify the filled orbital which has the higher tendency of donating electrons while lowest unoccupied molecular orbital connotes the empty orbitals which accept electrons [31]. Frontier molecular orbital analysis is used to assess the energy difference between the Highest Occupied Molecular Orbital (HOMO) and The Lowest Unoccupied Molecular Orbital (LUMO), Ionization potential (I=-EHOMO), electron affinity (A=-ELUMO), electronegativity (χ), global hardness (η), softness (S), electrophilicity, and dipole moment (ω). These descriptors are essential for predicting the chemical reactivity of potential drug candidates. As depicted in Table 1, Talimunoside I has the highest HOMO energy, followed by Varespladib, Kaempferol, Varespladib Methyl, Quercetin, Rutin, and Darapladib, respectively. The higher the HOMO energy the lower the ionization energy, therefore these results indicate that Talimunoside I and Varespladib possess low ionization energies, suggesting they have a strong tendency to act as good electron donors to the target protein when compared to rutin. Furthermore, the low LUMO (Lowest Unoccupied Molecular Orbital) energies of Varespladib (0.055eV) and Talimunoside I (0.051eV) further support their potential as electron acceptors.
Table 1:Physicochemical properties of top-scoring compounds.

Molecules with lower LUMO energies tend to be better electron acceptors. These findings align with the observed ionization potential and electron affinity values in Table 2. HOMO -LUMO energy gap is one of the critical indicators in predicting the interaction of drug candidate with target biomolecules. A smaller HOMO-LUMO gap generally indicates higher reactivity and lower stability [30]. When the gap is narrow, it’s easier for a molecule to lose an electron (oxidation in the HOMO) or gain an electron (reduction in the LUMO), making it more prone to reactions.
Table 2:Descriptors of absorption and distribution.

The results from the studies shows the value of HOMO-LUMO energy gap in increasing order; Talinumoside I<Varespladib<Quercetin<Kaempferol<Varespladib Methyl<Darapladib<Rutin. This trend suggests a corresponding difference in their reactivity. Drug candidate with a smaller HOMO-LUMO gap (like Talinumoside I) are likely to be more reactive than rutin. The electronegativity value gives insight to the relative ability of molecules to attract electron towards itself during chemical bonding and its overall polarity [32]. As depicted in Table 3, the electronegativity values (χ) suggest that the compounds are moderately polar, with values ranging from 0.0072 to 0.135. Darapladib, with the highest electronegativity value of 0.135eV compared to the other compounds, has the greatest potential to form strong bonds with target proteins. Energy gap determines the chemical hardness and softness of a molecule [33]. The larger the energy gap the harder the molecule and vice versa. As shown in Table 4, the drug candidate possess relatively moderate hardness and softness value hence are chemically moderately stable and reactive. Another key descriptor of reactivity is the dipole moment which measure the inhomogeneity of charge distribution and stability of drug candidate [34]. As shown in Table 5, darapladib exhibit high dipole moment of (0.123debye) compared to other ligands indicating it relative reactivity with the surrounding media. Furthermore, the calculated dipole moment reveal that all tested ligands are polar in nature, which can improve their solubility and absorption.
Table 3:Descriptors for pharmacokinetics.

Table 4:Excretion and toxicity parameters.

Table 5:Drug likeness and bioavailability score.

ADMET Investigation
The ADMET property of a drug candidate is part of the determining factor of its success and acceptability [35]. Safety issues can be contained with the use of computational ADMET [36]. The physicochemical profiling of the lead compounds in this study are shown in Table 6. The molecular weight ranged between 286.24 and 610.52g/mol with rutin and kaempferol having the highest and least values respectively.
Table 6:Illustration of the molecular orbitals of lead compounds and the standard compounds.

The Topological Polar Surface Area (TPSA) is essential for the prediction for absorption in intestine and brain. TPSA<60Å2 shows that the drug can penetrate the Blood Brain Barrier (BBB). Kaempferol and rutin having 111.13 and 269.43Å2 respectively as values of TPSA are the least and highest amongst the lead compounds. This confirms that all the four lead compounds do not penetrate the BBB and it is important so as not to induce an adverse drug reaction via the central nervous system. LogP is the term used to depict molecules’ lipophilicity and the value is determined through the estimation of the partitioning between phases that is aqueous and lipophilic [37]. In drug research, logP is an essential parameter because it has an influence on absorption, distribution, drug clearance route and permeability [37]. The logP value should be <5 and the most ideal values are between 1.35 and 1.8. Kaempferol, quercetin and talinumoside1 have logP values of 1.58, 1.23, and 1.80 respectively which are suitable values for drug candidates. If the logP is very low, the drug will not be retained and when too high, there will be a deposition of the drugs in fatty tissues. The logS shows water solubility level of individual molecule and this determines movement in hydrophilic state during distribution [38]. As shown in Table 7; Rutin, kaempferol and quercetin having logS values of -3.30, -3.31 and -3.16 respectively fall in the same range as varespladib and varespladib methyl which have their values as -3.80 and -4.01. Talinumoside1 with a value of -6.34 is significantly the same as Darapladib with a value of -7.75. These correlations show that the compounds have the potential of exhibiting similar hydrophilic property as the standards. P-glycoprotein (p-gp) is a transmembrane that expels numerous harmful compounds inside the cell to the extracellular space, but also has the ability to efflux many drugs out of the cells which can adversely affect the activity of diverse drugs [37]. P-gp substrates reduce drug absorption and therefore, the knowledge of its status is critical for drug development.
Table 7:Quantum chemical calculations of lead compound and the standard compound.

Kaempferol and quercetin are not substrates of P-gp and this depicts that the drug absorption level will be optimal. Rutin and talinumoside1 are P-gp substrate just as darapladib and as such the absorption rate will be minimal. Cytochrome P (CYP) enzymes are the most investigated phase 1 enzymes which plays the role of drug metabolism through the mediation of oxidation in numerous compounds [35]. Studies have shown that 75% of drugs in the market are metabolized by CYPs [39]. Rutin and talinumoside1 do not inhibit the activities of the CYP’s analyzed (which include CYP1A2, CYP2C19, CYP2C9, CYP2D6 and CYP3A4) but just as darapladib, kaempferol and quercetin can be an inhibitor to CYP1A2, CYP2D6 and CYP3A4 which may elicit drug-drug interaction. For these compounds, provisions should be made to enable the improvement of drug metabolism that might have been impeded. The analysis of the toxicity of these compounds showed that they are all not an inducer of hepatoxicity, carcinogenicity and cytotoxicity asides quercetin which has a minimal potential of being carcinogenic. Further studies can be performed to investigate the degree and the dose that may likely prompt this. It is worth noting that varespladib and varespladib methyl both have the potential of being cytotoxic while the later can also be carcinogenic. The oral LD50 of the quercetin, talinumoside1, kaempferol and rutin are 159, 3220, 3919 and 5000 (mg/kg). These values are needed to be known for the indication of the acute toxicity of the test compounds. Abbott Bioavailability score is a representation of the dose fraction that gets into system circulation after oral administration or via the extravascular route. This is an important parameter for drug absorption. The optimal score for bioavailability is ≥ 0.55 and this is the case for kaempferol and quercetin with both having a score of 0.55. Kaempferol and quercetin will have an ideal absorption rate. Scores of 0.11 and 0.17 possessed by talinumoside1 and rutin predicts poor oral bioavailability which is the same as darapladib. Kaempferol and quercetin obeyed all rules of Lipinski, Egan and Veber. Rutin and talinumoside1 violated 3 rules of Lipinski and 1 of the rules of Egan and Veber. For the lipinski violation, it is due to the molecular weight being >500Da and this can be solved through lead optimization according to [38]. Also, the hydrogen bond donors and hydrogen bond acceptors were greater than 5 and 10 respectively. For the Egan and Veber rule violation, it was due to the TPSA being greater than 131.6 and 140 (respectively) for the compounds.
In conclusion, Talinum paniculatum has been used for folk medicine and studied to treat several health challenges due to its phytoconstituents and this prompted the use of drug discovery tools to screen bioactive compounds of the plant against PLA2 and metalloproteinase which are major enzymes present in venom. The lead compounds analyzed showed a very good binding affinity and binding energy in comparison to the standards. Kaempferol and quercetin had a good ADMET result but rutin and talinumoside1 can undergo modifications (such as lead optimization) to make them suitable drug candidates. The compounds may be explored as antivenins individually or in combination for the treatment of envenomation.
This study identified bioactive compounds from Talinum paniculatum with significant inhibitory potential against phospholipase A2 and metalloproteinases, the major enzymatic mediators of venom toxicity. Among the 40 compounds screened, quercetin and kaempferol emerged as the most promising candidates, demonstrating high binding affinities, favourable MM/ GBSA binding energies, and desirable pharmacokinetic properties compared with reference inhibitors such as varespladib and darapladib. Although rutin and talinumoside I also exhibited strong inhibitory activity, their pharmacokinetic limitations suggest that structural optimisation may be required for clinical application. Collectively, these findings highlight T. paniculatum as a valuable source of plant-derived antivenom leads, warranting further in vitro and in vivo validation for therapeutic development.
References
- Ahmadi S, Knerr JM, Argemi L, Bordon KC, Pucca MB, et al. (2020) Scorpion venom: Detriments and benefits. Biomedicines 8(5): 118.
- Ismail M (1995) The scorpion envenoming syndrome. Toxicon 33(7): 825-858.
- Zhang XY, Zhang PY (2020) Scorpion venoms in gastric cancer. Oncology Letters 20(5): 3683-3686.
- Scieuzo C, Salvia R, Franco A, Pezzi M, Cozzolino F, et al. (2021) An integrated transcriptomic and proteomic approach to identify the main Torymus sinensis venom components. Scientific Reports 11(1): 5032.
- Isbister GK (2010) Snakebite doesn't cause disseminated intravascular coagulation: Coagulopathy and thrombotic microangiopathy in snake envenoming. Seminars in Thrombosis and Hemostasis 36(4): 444-451.
- Herrera M, Fernández J, Vargas M, Villalta M, Segura Á, et al. (2012) Comparative proteomic analysis of the venom of the taipan snake, Oxyuranus scutellatus, from Papua New Guinea and Australia: Role of neurotoxic and procoagulant effects in venom toxicity. Journal of Proteomics 75(7): 2128-2140.
- Gulati A, Isbister GK, Duffull SB (2013) Effect of Australian elapid venoms on blood coagulation: Australian Snakebite Project (ASP-17). Toxicon 61: 94-104.
- O’Rourke KM, Correlje E, Martin CL, Robertson JD, Isbister GK (2013) Point-of-care derived INR does not reliably detect significant coagulopathy following Australian snakebite. Thrombosis Research 132(5): 610-613.
- Incamnoi P, Patramanon R, Thammasirirak S, Chaveerach A, Uawonggul N, et al. (2013) Heterotonic (HmTx), a novel heterodimeric phospholipase A2 from Heterometrus laoticus scorpion venom. Toxicon 61: 62-71.
- Six DA, Dennis EA (2000) The expanding superfamily of phospholipase A2 enzymes: Classification and characterization. Biochim Biophysica Acta (BBA) 1488(1-2): 1-19.
- Bulfone TC, Samuel SP, Bickler PE, Lewin MR (2018) Developing small molecule therapeutics for the initial and adjunctive treatment of snakebite. Journal of Tropical Medicine 4320175.
- Fry B (2015) Venomous reptiles and their toxins: Evolution, pathophysiology and biodiscovery. Oxford University Press, New York, USA, pp. 606.
- Fox JW, Serrano SM (2008) Insights into and speculations about Snake Venom Metalloproteinase (SVMP) synthesis, folding and disulfide bond formation and their contribution to venom complexity. The FEBS Journal 275(12): 3016-3030.
- Fox JW, Serrano SM (2005) Structural considerations of the snake venom metalloproteinases, key members of the M12 reprolysin family of metalloproteinases. Toxicon 45(8): 969-985.
- Snyder DW, Bach NJ, Dillard RD, Draheim SE, Carlson DG, et al. (1999) Pharmacology of LY315920/S-5920, [[3-(Aminooxoacetyl)-2-ethyl-1-(phenylmethyl)-1H-indol-4-yl] oxy] acetate, a potent and selective secretory phospholipase A2 inhibitor: A new class of anti-inflammatory drugs, SPI. Journal of Pharmacology and Experimental Therapeutics 288(3): 1117-1124.
- Lewin M, Samuel S, Merkel J, Bickler P (2016) Varespladib (LY315920) appears to be a potent, broad-spectrum, inhibitor of snake venom phospholipase A2 and a possible pre-referral treatment for envenomation. Toxins 8(9): 248.
- Tyagi MG, Vyas DJ, Mukherjee VE (2018) Development of novel phospholipase A2 inhibitors using molecular and computational techniques. IOSR-JPBS 13(2): 6-12.
- Reis LF, Cerdeira CD, Paula BF, Silva JJ, Coelho LF, et al. (2015) Chemical characterization and evaluation of antibacterial, antifungal, antimycobacterial, and cytotoxic activities of Talinum paniculatum. Rev Inst Med Trop Sao Paulo 57(5): 397-405.
- Coelho FC, Tirloni CA, Marques AA, Gasparotto FM, Lívero FA, et al. (2019) Traditional plants used by remaining healers from the region of Grande Dourados, Mato Grosso do Sul, Brazil. Journal of Religion and Health 58(2): 572-588.
- Holford M, Daly M, King GF, Norton RS (2018) Venoms to the rescue. Science 361(6405): 842-844.
- Hou T, Wang J, Li Y, Wang W (2011) Assessing the performance of the MM/PBSA and MM/GBSA methods. 1. The accuracy of binding free energy calculations based on molecular dynamics simulations. Journal of Chemical Information and Modeling 51(1): 69-82.
- Neves MA, Totrov M, Abagyan R (2012) Docking and scoring with ICM: The benchmarking results and strategies for improvement. Journal of Computer-Aided Molecular Design 26(6): 675-686.
- Gandini D, Gogioso L, Bolognesi M, Bordo D (1996) Patterns in ionizable side chain interactions in protein structures. Proteins: Structure, Function, and Bioinformatics 24(4): 439-449.
- Barril X, Aleman C, Orozco M, Luque FJ (1998) Salt bridge interactions: Stability of the ionic and neutral complexes in the gas phase, in solution, and in proteins. Proteins: Structure, Function, and Bioinformatics 32(1): 67-79.
- Chen D, Oezguen N, Urvil P, Ferguson C, Dann SM, et al. (2016) Regulation of protein-ligand binding affinity by hydrogen bond pairing. Science Advances 2(3): e1501240.
- Zhang Z, Chen H, Bai H, Lai L (2007) Molecular dynamics simulations on the oligomer-formation process of the GNNQQNY peptide from yeast prion protein Sup35. Biophysical Journal 93(5): 1484-1492.
- Hunter CA, Singh J, Thornton JM (1991) π-π interactions: The geometry and energetics of phenylalanine-phenylalanine interactions in proteins. Journal of Molecular Biology 218(4): 837-846.
- Myslinski JM, Clements JH, Martin SF (2014) Protein-ligand interactions: Probing the energetics of a putative cation-π Bioorganic and Medicinal Chemistry Letters 24(14): 3164-3167.
- Mamy L, Patureau D, Barriuso E, Bedos C, Bessac F, et al. (2015) Prediction of the fate of organic compounds in the environment from their molecular properties: A review. Critical Reviews in Environmental Science and Technology 45(12): 1277-1377.
- Miar M, Shiroudi A, Pourshamsian K, Oliaey AR, Hatamjafari F (2020) Theoretical investigations on the HOMO–LUMO gap and global reactivity descriptor studies, natural bond orbital, and nucleus-independent chemical shifts analyses of 3-phenylbenzo [d] thiazole-2 (3 H)-imine and its para-substituted derivatives: Solvent and substituent effects. Journal of Chemical Research 45(1-2):147-158.
- Hagar M, Ahmed HA, Aljohani G, Alhaddad OA (2020) Investigation of some antiviral N-heterocycles as COVID 19 drug: Molecular docking and DFT calculations. International Journal of Molecular Sciences 21(11): 3922.
- Sproul GD (2020) Evaluation of electronegativity scales. ACS Omega 5(20): 11585-11594.
- Balogun TA, Ipinloju N, Abdullateef OT, Moses SI, Omoboyowa DA, et al. (2021) Computational evaluation of bioactive compounds from Colocasia affinis Schott as a novel EGFR inhibitor for cancer treatment. Cancer Informatics 20: 11769351211049244.
- Puranen JS, Vainio MJ, Johnson MS (2010) Accurate conformation‐dependent molecular electrostatic potentials for high‐throughput in silico drug discovery. Journal of Computational Chemistry 31(8):1722-1732.
- Moroy G, Martiny VY, Vayer P, Villoutreix BO, Miteva MA (2012) Toward in silico structure-based ADMET prediction in drug discovery. Drug Discovery Today 17(1-2): 44-55.
- Merlot C (2010) Computational toxicology-a tool for early safety evaluation. Drug Discovery Today 15(1-2): 16-22.
- Chandrasekaran B, Abed SN, Attraqchi AO, Kuche K, Tekade RK (2018) Computer-aided prediction of pharmacokinetic (ADMET) properties. Dosage Form Design Parameters, Academic Press, Massachusetts, USA, pp. 731-755.
- Akinnusi PA, Olubode SO, Alade AA, Ashimi AA, Onawola OL, et al. (2023) Potential inhibitory biomolecular interactions of natural compounds with different molecular targets of diabetes. Bioinformatics and Biology Insights 17: 11779322231167970.
- Guengerich FP (2008) Cytochrome p450 and chemical toxicology. Chemical Research in Toxicology 21(1): 70-83.
© 2025 Bakare OS. This is an open access article distributed under the terms of the Creative Commons Attribution License , which permits unrestricted use, distribution, and build upon your work non-commercially.
Editor In Chief







.jpg)

























Signup for Newsletter

Quick Links
 Editorial Board Registrations
Editorial Board Registrations Submit your Article
Submit your Article Refer a Friend
Refer a Friend Advertise With Us
Advertise With UsOur Recent Edition

.jpg)



Top Editors




.jpg)

















.bmp)
.jpg)
.png)
.jpg)













.jpg)






.png)

.png)




.png)




Financial Support

Sponsors

Latest e-Books

Latest Video

 a Creative Commons Attribution 4.0 International License. Based on a work at www.crimsonpublishers.com.
Best viewed in
a Creative Commons Attribution 4.0 International License. Based on a work at www.crimsonpublishers.com.
Best viewed in