
- About Us
- Information
-

The Author ensures that the research has been conducted responsibly and ethically with adherence to all relevant regulations. read more..
- For Authors
- For Reviewer
- Manuscript Guidelines
- Membership
- Publication Ethics
-
- Journals
- Reprints
- e-Books
- Videos
- Policies
- Contact Us
 COVID-19
COVID-19
- Submissions



Full Text
Research & Development in Material Science
Modeling of Cr3+ Doped β-Ga2O3 Single Crystals
Maroj Bharati1, Vikram Singh1 and Ram Kripal2*
1Department of Physics, Nehru Gram Bharti (DU), India
2EPR Laboratory, Department of Physics, University of Allahabad, India
*Corresponding author:Ram Kripal, EPR Laboratory, Department of Physics, University of Allahabad, Prayagraj-211002, India
Submission: September 14, 2023;Published: October 02, 2023

ISSN: 2576-8840 Volume 19 Issue 3
Abstract
Crystal field parameters and zero field splitting parameters of Cr3+ doped β-Ga2O3 single crystals are computed with the help of superposition model. The appropriate sites for Cr3+ ions in β-Ga2O3 with distortion are adopted for calculation. The experimental values of zero field splitting parameters are in good match with the theoretical ones when local distortion is included into computation. The optical energy band positions for Cr3+ in β-Ga2O3 are calculated using crystal field parameters and Crystal Field Analysis Program. The results indicate that Cr3+ ions substitute at GaII3+ octahedral sites in β-Ga2O3 single crystals.
Keywords: Superposition model; Crystal field; Zero-field splitting; Optical spectroscopy; Cr3+ ions in β-Ga2O3
Opinion
Electron Paramagnetic Resonance (EPR) is a powerful tool for investigating the local site symmetry and Zero Field Splitting (ZFS) of impurity ions in host crystals [1,2]. Superposition Model (SPM) [3-5] is frequently employed to find theoretically the Crystal Field (CF) parameters and Zero Field Splitting (ZFS) parameters.
β-Ga2O3, gallium oxide, is an insulator having a band gap of 4.8eV [6]. When synthesized under reducing conditions, it becomes an n-type semiconductor [7]. Doping yields variation in the conductivity of both the p and n types. These materials have potential applications in optoelectronics These are also used as insulating or conductive window material or substrates [8]. As the above material is stable at high temperatures, it has been widely studied for application as a gas sensor [9]. The electrical conductivity of Ga2O3 at large temperatures is changed in the presence of oxidizing or reducing gases. β-Ga2O3 has become a good material for optoelectronic devices in the deep ultraviolet region [10]. Gallium oxide crystal doped with trivalent transition ions is of importance in microwave, optical maser and exchange interaction between substitutional ions studies [11]. Introduced impurities giving microscopic structural change affect the optical properties of the crystal based on location of the impurities. The Cr3+ion is a very important probe for obtaining information on β-Ga2O3 crystal.
The EPR study of Cr3+ doped β-Ga2O3 has been reported [12]. The EPR spectra of Cr3+ ions in β-Ga2O3 single crystals were recorded on an X-band spectrometer at room temperature. These spectra in the three mutually perpendicular planes were analyzed employing an effective spin Hamiltonian. The spectroscopic splitting factor g and the Zero Field Splitting (ZFS) parameters of Cr3+ ions were obtained for the octahedral substitutional sites in β-Ga2O3 single crystal. The actual local site symmetry of Cr3+ ions in the crystal was also investigated.
The laboratory axes (x, y, z) are chosen to be along the crystallographic axes (a, b, c*). The symmetry adopted axes (magnetic axes) are labeled (X, Y, Z). The principal Y axis of g and D tensors of Cr3+ ions is found parallel to the crystallographic b axis (monoclinic C2 axis). The principal axes of g and D tensors in the plane normal to the C2 axis do not coincide. If an external magnetic field is applied in a plane not containing the C2 axis, the resonance fields are asymmetric about the extrema [12]. These principal directions and the asymmetry show that the local site symmetry of Cr3+ ions in β-Ga2O3 crystal is monoclinic. In the principal magnetic axis system, the parameters g, D and E were obtained using EPR-NMR program. There are two chemically distinguishable Ga3+ sites in the unit cell: Ga13+ ions coordinated with four oxygens and Ga23+ ions coordinated with six oxygens [13]. The Cr3+ ion EPR spectrum shows a single set of three lines indicating Cr3+ ions to be located at only one site. Cr3+ ions with an ionic radius 0.0615nm for the six-fold coordination prefer the octahedrally coordinated Ga23+ site (ionic radius 0.062nm) [12].
The present investigation presents the Superposition Model (SPM) of the CF parameters and the ZFS parameters for Cr3+ ions in β-Ga2O3 crystal. The aim is to obtain the ZFS parameters, the CF parameters and the distortion in the lattice for the Cr3+ ions in β-Ga2O3 at octahedral sites. The optical energy band positions for Cr3+ ions in β-Ga2O3 are computed using CF parameters and Crystal Field Analysis (CFA) program. The CF parameters and ZFS parameters evaluated may be useful in future investigations for scientific and industrial applications of such crystals.
Crystal structure
The crystal structure of β-Ga2O3 is monoclinic with space group C2/m [14]. The lattice parameters are a =1.2214nm, b=0.30371nm, c=0.57981nm, β=103.83° and Z=4. Two chemically distinct cation sites are coordinated with oxygens either tetrahedrally or octahedrally. The structure shows double chains of GaO6 octahedra (Ga2) parallel to the b axis, which are connected by GaO4 tetrahedra (Ga1). The crystal structure of β-Ga2O3 with symmetry adopted axis system (SAAS) is given in Figure 1.
Figure 1:Crystal structure of β-Ga2O3 with symmetry adopted axis system (SAAS).

The Symmetry Adopted Axes (SAA) (local site symmetry axes) are the mutually perpendicular directions of metal-ligand bonds. The Z axis of SAAS is along the metal-ligand bond Ga2-O1 (crystal c*-axis) and the two other axes (X,Y) are perpendicular to the Z axes (in the ab plane) for center I (Figure 1). This shows that Cr3+ substitutes for Ga23+ in the crystal of β-Ga2O3 with approximately orthorhombic symmetry. The ionic radius of Cr3+ ion (0.0615nm) [15] is slightly smaller than the ionic radius of Ga23+ (0.062nm) suggesting Cr3+ ion to sit at the location of Ga23+ with certain distortion.
The Cr3+ ion position and spherical polar coordinates of the ligands in β-Ga2O3 [14] for center I are given in Table 1. These data are used for ZFS and CF calculations for Cr3+ ion in β-Ga2O3.
Table 1:Fractional coordinates of Cr3+ ion (center I) and spherical co-ordinates (R, θ, ф) of ligands in β-Ga2O3 single crystal.

Calculations of zero field splitting parameters
The spin Hamiltonian [16-18] used to find the energy levels of Cr3+ ions in crystals is:
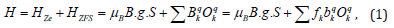
Where g, μB and B are the spectroscopic splitting factor, Bohr magneton and static magnetic field, respectively. S represents the effective spin operator and Oqk(Sx,Sy,Sz) are the Extended Stevens Operators (ESO) [19,20]; Bqk and bqk are the ZFS parameters, ƒk=1/3 and 1/60 are the scaling factors for k = 2 and 4, respectively. The ZFS terms in (1) for Cr3+ ion (S=3/2) at orthorhombic symmetry sites are given as [21,22]:
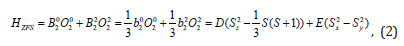
The conventional orthorhombic ZFS parameters D, E and Bqk are related as follows:
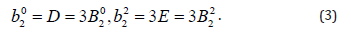
The ZFS parameters (in ESO notation) for any symmetry employing SPM [21,22] are obtained as:
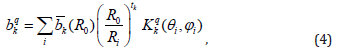
Where (Ri, θI, ϕi) are the spherical polar coordinates of i-th ligand. The intrinsic parameters bk provide the strength of the k-th rank ZFS contribution from a ligand at the distance Ri and the coordination factors Kqk give the geometrical information. Kqk for k = 1 to 6 in the ESO notation [23] are presented in Appendix A1 of [24].
Eq. (4) provides conventional ZFS parameters, D and E, in terms of the intrinsic parameters bk , the power-law exponents tk, and the reference distance R0, as [24,25-27]:
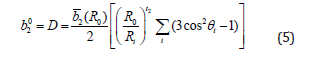
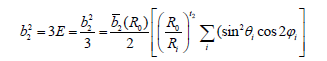
Cr3+ ion in β-Ga2O3 may be supposed to substitute at the Ga23+ ion site, and the interstitial site with similar ligand arrangement. The local symmetry at Cr3+ ion site is assumed to be approximately orthorhombic. In octahedral coordination of Cr3+ ion in LiNbO3 having Cr3+-O2- bond, 2 0 b (R )=2.34cm-1 and t2=-0.12 [28] were used to obtain b02 and b22 . Because Cr3+ ion in β-Ga2O3 has distorted octahedral coordination (Figure 1) with oxygen as ligands, the bqk in the present investigation are determined using b2 (R0) =2.34cm- 1 and t2=-1.96 for the center I.
The Cr3+ ion position and spherical coordinates of ligands shown in Table 1 are used for calculation. The conventional ZFS parameters, D and E of Cr3+ ion in β-Ga2O3 single crystal are evaluated employing Eq. (5). The reference distance R0=0.200nm was taken for the calculation of ZFS parameters [29], and the values are: |D|=88.7×10-4cm-1 and |E|=25.7×10-4 cm-1 for center I. The ratio b22/b02 should be within the range (0, 1) for orthorhombic symmetry [30]. In the present computation, the ratio 2 0 |b22|/|02| =0.870 and |E| / |D| = 0.290 for center I. It is seen that the value of |D| and |E| do not agree with the experimental values though |b22|/|b02| is in the specified range [30]. Hence, with above t2 and reference distance R0, the ZFS parameters |D| and |E| are calculated for Cr3+ at the Ga23+ site with distortion having position Ga23+ (0.49350, 0.55400, 1.15092) for center I. The conventional ZFS parameters found now are |D|=5385.2×10-4cm-1, |E|=1288.2×10- 4cm-1 for center I, which are in good match with the experimental ones. The ratio |b22|/|b02| =0.717 and |E| / |D| = 0.239 for center I are in the specified range [30]. Further, with above t2 and reference distance R0, the ZFS parameters |D| and |E| are computed for Cr3+ at the interstitial site but the values found in this case are inconsistent with the experimental values. Hence, these data are not shown here.
The calculated and experimental ZFS parameters for Cr3+ ion in β-Ga2O3 are given in Table 2. It is seen from Table 2 that the ZFS parameters |D| and |E| are in good match with the experimental ones [12] when the distortion is introduced into calculation.
Table 2:Calculated and experimental ZFS parameters of Cr3+ doped β-Ga2O3 single crystal for center I along with reference distance.

ND = No distortion, WD = With distortion, e = experimental.
Calculations of crystal field parameters
The CF energy levels of transition ions in crystals [31-34] using Wybourne operators [15,16,35] are given by:
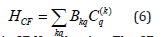
Where ℋ CF is CF Hamiltonian. The CF parameters in (6) for a metal-ligand complex are determined using SPM [21,22] as:
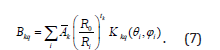
Where R0 is the reference distance; Ri, θi, φi are the spherical polar coordinates of the ith ligand and Kkq are the coordination factors [31]. To obtain Bkq (k=2, 4; q=0, 2, 4); A = 40, 400cm-1, t2 =1.3, 4 A =11, 700cm-1 and t4 = 3.4 are used [31]. The calculated Bkq parameters are presented in Table 3. The ratio B22/B02 = 0.255 for center I, which indicates that Bkq parameters are standardized [30]. Taking Bkq parameters given in Table 3 and CFA program [32,33], the CF energy levels of Cr3+ ion in β -Ga2O3 single crystals are calculated by diagonalizing the complete Hamiltonian. The calculated energy values are given in Table 4. The calculated energy values are compared with the experimental energy values for Cr3+: β-Ga2O3 [36]. From Table 4, it is noted that the theoretical and experimental band positions are in reasonable match. Thus the theoretical study of Cr3+ ions at Ga23+ sites in β-Ga2O3 supports the experimental one [12,36].
Table 3:Bkq parameters of Cr3+ doped β-Ga2O3 single crystal for center I with distortion.

WD = With distortion.
Table 4:Experimental and calculated energy band positions (centers I) of Cr3+ doped β-Ga2O3 single crystal.

(Racah parameters A, B and C, spin-orbit coupling constant and Trees correction are 0, 726.3, 2905.2 (= 4B), 276 and 70cm-1, respectively)
The optical absorption/excitation spectra of Cr3+-activated phosphors have been explained with the help of Franck-Condon analysis with the Configurational-Coordinate (CC) model [37]. The different excited state-ground state transitions in Cr3+ are because of the strong coupling with the lattice vibrations (CC model) [37]. The CC model has not been employed and hence there is difference between excited-state peak energies found here and Zero-Phonon Line (ZPL) energies discussed in [37,38]. The Cr3+-activated oxide phosphors are classified into two groups: O-Cr-A and O-Cr-B [38]. Phosphors of O-Cr-A type have an energy inequality relation of E(2Eg)ZPL < E(4T2g)ZPL, giving a series of the sharp emission peaks by the 2Eg → 4A2g transitions. Phosphors of O-Cr-B type have an energy inequality relation of E(4T2g) ZPL < E(2Eg) ZPL, giving a broad emission band due to the 4T2g → 4A2g transitions. β-Ga2O3: Cr3+ comes under O-Cr-B type phosphors [38].
Summary and Conclusion
The Zero-Field Splitting (ZFS) parameters and Crystal Field (CF) parameters for Cr3+ ions in β -Ga2O3 single crystals are calculated with the help of Superposition Model (SPM). Cr3+ ions in β -Ga2O3 crystal at Ga23+ ion sites, interstitial site and distortion models are considered for calculation. The calculated conventional ZFS parameters for Cr3+ ion at Ga23+ sites in β-Ga2O3 single crystal provide good agreement with the experimental values when distortion is included in the calculation. It is noted that the Cr3+ ions substitute at Ga23+ ion sites in β-Ga2O3 lattice. The CF energy values for Cr3+ ions at Ga23+ sites computed using CFA package and CF parameters are in reasonable match with the experimental ones. Thus, the theoretical results support the experimental study.
Modeling procedure employed in this investigation may be useful in future to correlate EPR and optical data for different ion-host systems in exploring crystals for various scientific and industrial applications.
Acknowledgement
The authors are thankful to the Head, Department of Physics for providing the departmental facilities and to Prof. C. Rudowicz, Faculty of Chemistry, A. Mickiewicz. University, Poznan, Poland for CFA program.
References
- Mabbs FE, Collison D, Gatteschi D (1992) Electron paramagnetic resonance of d transition metal compounds. Elsevier, Amsterdam, Netherlands.
- Weil JA, Bolton JR (2007) Electron paramagnetic resonance: elementary theory and practical applications. (2nd edn), Wiley, New York, USA.
- Brik MG, Avram CN, Avram NM (2006) Calculations of spin Hamiltonian parameters and analysis of trigonal distortions in LiSr(Al,Ga)F6:Cr3+ Physica B 384(1-2): 78-81.
- Pandey S, Kripal R, Yadav AK, Açıkg¨oz M, Gnutek P, et al. (2021) Implications of direct conversions of crystal field parameters into zero-field splitting ones - Case study: Superposition model analysis for Cr3+ ions at orthorhombic sites in LiKSO4. J Lumin 230: 117548.
- Bradbury MI, Newman DJ (1967) Ratios of crystal field parameters in rare earth salts. Chem Phys Lett 1(2): 44-45.
- Tippins HH (1965) Optical absorption and photoconductivity in the Bandedge of β-Ga2O3. Phys Rev 140: A316-A319.
- Hajnal Z, Miro J, Kiss G, Reti F, Deak P, et al. (1999) Role of oxygen vacancy defect states in the n-type conduction of β-Ga2O3. J Appl Phys 86: 3792-3796.
- Tomm Y, Ko JM, Yoshikawa A, Fukuda T (2002) Floating zone growth of β-Ga2O3: A new window material for optoelectronic device applications. Sol Energy Mater Sol Cells 66: 369-374.
- Grank J, Fleischer M, Meixner H (1996) Electrical doping of gas-sensitive, semiconducting Ga2O3 thin films. Sens Actuators B 34: 373-377.
- Orita M, Ohta H, Hirano M, Hosono H (2000) Deep-ultraviolet transparent conductive β-Ga2O3 thin films. Appl Phys Lett 77: 4166-4168.
- Gesmundo F, De Asmundis C (1973) An electron paramagnetic resonance study of the solid solutions of chromia in β-Ga2O3. J Phys Chem Solids 34: 1757-1763.
- Yeom TH, Kim IG, Lee SH, Choh SH, Yu YM (2003) Electron paramagnetic resonance characterization of Cr3+ impurities in a β-Ga2O3 single crystal. J Appl Phys 93: 3315-3319.
- Geller S (1960) Crystal structure of β-Ga2O3. J Chem Phys 33: 676-684.
- Ahman J, Svensson G, Albertsson J (2000) A reinvestigation of β~-gallium oxide. Acta Cryst C 52(6): 1336-1338.
- Figgis BN, Hitchman MA (2000) Ligand field theory and its applications, Wiley, New York, USA.
- Rudowicz C, Karbowiak M (2015) Disentangling intricate web of interrelated notions at the interface between the physical (crystal field) Hamiltonians and the effective (spin) Hamiltonians. Coord Chem Rev 287: 28-63.
- Rudowicz C (1987) Concept of spin hamiltonian, forms of zero field splitting and electronic zeeman hamiltonians and relations between parameters used in EPR: A critical review. Magn Reson Rev 13: 1-89.
- Rudowicz C, Misra SK (2001) Spin-Hamiltonian formalisms in Electron Magnetic Resonance (EMR) and related spectroscopies. Appl Spectrosc Rev 36(1): 11-63.
- Rudowicz C (1985) Transformation relations for the conventional Okq and normalised O'kq Stevens operator equivalents with k=1 to 6 and –k≤q≤ J Phys C Solid State Phys 18(7): 1415-1430.
- Rudowicz C, Chung CY (2004) The generalization of the extended Stevens operators to higher ranks and spins, and a systematic review of the tables of the tensor operators and their matrix elements. J Phys Condens Matter 16(32): 5825-5847.
- Newman DJ, Ng B (2000) Superposition model. In: Newman DJ, Ng B (Eds.), Crystal Field Handbook, Cambridge University Press, UK, pp. 83-119.
- Newman DJ, Ng B (1989) The superposition model of crystal fields. Rep Prog Phys 52: 699-763.
- Rudowicz C (1987) On the derivation of the superposition-model formulae using the transformation relations for the Stevens operators. J Phys C: Solid State Phys 20(35): 6033-6037.
- Rudowicz C, Gnutek P, Açıkgöz M (2019) Superposition model in electron magnetic resonance spectroscopy – a primer for experimentalists with illustrative applications and literature database. Appl Spectroscopy Rev 54: 673-718.
- Açıkgöz M (2012) A study of the impurity structure for 3d3 (Cr3+ and Mn4+) ions doped into rutile TiO2 Spectrochim Acta A 86(2): 417-422.
- Müller KA, Berlinger W, Albers J (1985) Paramagnetic resonance and local position of Cr3+ in ferroelectric BaTiO3. Phys Rev B 32(9): 5837-5850.
- Müller KA, Berlinger W (1983) Superposition model for sixfold-coordinated Cr3+ in oxide crystals (EPR study). J Phys C: Solid State Phys 16(35): 6861-6874.
- Yeom TH, Chang YM, Rudowicz C (1993) Cr3+ centres in LiNbO3: Experimental and theoretical investigation of spin Hamiltonian parameters. Solid State Commun 87(3): 245-249.
- Siegel E, Muller KA (1979) Structure of transition-metal-oxygen-vacancy pair centers. Phys Rev B 19(1): 109-120.
- Rudowicz C, Bramley R (1985) On standardization of the spin Hamiltonian and the ligand field Hamiltonian for orthorhombic symmetry. J Chem Phys 83(10): 5192-5197.
- Yeung YY, Newman DN (1986) Superposition-model analyses for the Cr3+ 4A2 ground state. Phys Rev B 34(4): 2258-2265.
- Yeung YY, Rudowicz C (1992) Ligand field analysis of the 3dN ions at orthorhombic or higher symmetry sites. Comp Chem 16(3): 207-216.
- Yeung YY, Rudowicz C (1993) Crystal field energy levels and state vectors for the 3dn ions at orthorhombic or higher symmetry sites. J Comput Phys 109(1): 150-152.
- Chang YM, Rudowicz C, Yeung YY (1994) Crystal field analysis of the 3dN ions at low symmetry sites including the ‘imaginary’ terms. Computers in Physics 8(5): 583-588.
- Wybourne BG (1965) Spectroscopic properties of rare earth. Wiley, New York, USA.
- Zhang JG, Bin L, Tai XC, Jun X, Qun D, et al. (2007) Single crystal β-Ga2O3: Cr grown by floating zone technique and its optical properties. Sci China Ser E-Tech Sci 50(1): 51-56.
- Adachi S (2019) Photoluminescence spectroscopy and crystal-field parameters of Cr3+ ion in red and deep red-emitting phosphors. ECS J Solid State Sci Tech 8(12): R164-R168.
- Adachi S (2021) Review-photoluminescence properties of Cr3+-activated oxide phosphors. ECS J Solid State Sci Tech 10: 026001.
© 2023 Ram Kripal. This is an open access article distributed under the terms of the Creative Commons Attribution License , which permits unrestricted use, distribution, and build upon your work non-commercially.
Editor In Chief







.jpg)

























Signup for Newsletter

Quick Links
 Editorial Board Registrations
Editorial Board Registrations Submit your Article
Submit your Article Refer a Friend
Refer a Friend Advertise With Us
Advertise With UsOur Recent Edition

.jpg)



Top Editors




.jpg)

















.bmp)
.jpg)
.png)
.jpg)













.jpg)






.png)

.png)




.png)




Financial Support

Sponsors

Latest e-Books

Latest Video

 a Creative Commons Attribution 4.0 International License. Based on a work at www.crimsonpublishers.com.
Best viewed in
a Creative Commons Attribution 4.0 International License. Based on a work at www.crimsonpublishers.com.
Best viewed in