
- About Us
- Information
-

The Author ensures that the research has been conducted responsibly and ethically with adherence to all relevant regulations. read more..
- For Authors
- For Reviewer
- Manuscript Guidelines
- Membership
- Publication Ethics
-
- Journals
- Reprints
- e-Books
- Videos
- Policies
- Contact Us
 COVID-19
COVID-19
- Submissions



Full Text
Polymer Science: Peer Review Journal
Synthesis of Oligo(Ethylene Glycol)- Functionalized Pyrroles and their Electrochemical Deposition
Wolfdietrich Meyer1*, Paul Seefeldt2 and Jens Schönewerk3
1Fraunhofer Institute for Applied Polymer Research IAP, Germany
2University of Applied Sciences Berlin, Germany
3Se ma Gesellschaft für Innovations mbH, Germany
*Corresponding author:Wolfdietrich Meyer, Fraunhofer Institute for Applied Polymer Research IAP, Potsdam Science Park, Geiselbergstrasse 69, 14476 Potsdam Germany
Submission: December 09, 2023;Published: January 05, 2024

ISSN: 2770-6613 Volume5 Issue3
Abstract
Four N-substituted pyrrole derivatives with oligo (ethylene glycol)-functionalization were synthesized and characterized by 1H and 13C NMR and FTIR. Namely 2-[2-(1H-Pyrrole-1-yl)-ethoxy]-ethan- 1-ol, 1-{17-[(tetrahydro-2H-pyran-2-yl)oxy]-3,6,9,12,15-pentaoxaheptadecyl}-1H-pyrrole, 1,1’- bis(2-(2-(2 ((tetrahydro-2H-pyran-2-yl)oxy)ethoxy)ethoxy)ethyl)-1H,1’H-2,2’-bipyrrole and 1-(2,3,5,6,8,9,11,12,14,15-decahydrobenzo[b][1,4,7,10,13,16]hexaoxacyclooctadecin-18yl)-1H-pyrrole were electrodeposited from acetonitrile electrolytes by cyclic voltammetry on a platinum electrode. The deposited films were characterized by its electro polymerizable behaviour and deposited masses calculated from the evaluated charges of the reversible redox peaks of the polypyrrole systems. Overoxidation of the polypyrrole redox system potentials were determined and Li+ ion conductivity within the deposited film was determined by impedance spectroscopy.
Keywords:Synthesis; Pyrrole; Electrochemical deposition
Introduction
The modification of electrodes with polypyrrole (PPy) is prevalent due to its high electric conductivity and chemical stability. These characteristics render it suitable for applications in various fields such as electronics, electrodes, batteries and supercapacitors, and sensors [1-7]. Polypyrroles versatility lies in its electrical conductivity, allowing it to function as a carrier material for numerous sensors, especially in biological systems where it is used as an enzyme, immuno, or DNA sensor. The utility of polypyrrole extends to the manufacture of organic light-emitting diodes and organic field-effect transistors. PPy is the most studied material among the conducting polymers for high-performance supercapacitors [8,9] and as described by Yadav et al. and addresses morphology collapse and poor cycling performances [10].
A crucial aspect of polypyrrole application is corrosion protection, with increasing efforts to replace chromium with polypyrrole. Compared to other electrically conductive polymers, polypyrrole stands out due to its water solubility, ease of oxidation, and superior environmental stability [11,12]. PPy has found use in lithium-ion batteries, addressing manufacturing convenience, over-coming the low specific capacity of PPy as a cathode material by integrating it with a ferrocene or nitrile functionality, and improving adhesive properties to electrodes via simultaneous electrochemical deposition of PPy with dopamine [2], overcoming low specific capacity of PPy as cathode material by combining it with a ferrocene functionality [4] or nitrile functionality to improve capacity retention when using LNC cathode materials [13], the adhesive properties to electrodes were improved by simultaneously electrochemical deposition of PPy with dopa-mine [14]. The PPy as electric conducting compound in the cathodes improves the electric conducting properties of the active materials and as we expect, the use of structurally extended PPy to OEG-PPy also improves the ionic conductivity. Overoxidized polypyrrole (oPPy) is a form of polypyrrole that has undergone overoxidation. Overoxidation of polypyrrole can change the properties of polypyrrole, making it e.g. permselective [15]. This means that over-oxidized polypyrrole can selectively allow certain ions to pass through while blocking others. This property can be useful in various applications, such as sensors and separation processes. For example, a convenient “one step” preparation process of molecularly imprinted overoxidizedpolypyrrole (oPPy) colloids by chemical polymerization has been described, which exhibits high uptake ability and enantioselectivity [16].
Here we address the synthesis of N-modified oligo(ethylene glycol) pyrrolderivative for the means of electrochemical deposition issues on electrodes as a tool to modify or cover up electrodes (as shown in Figure 1, thus describing possible application scenarios) with a PEG functionality which is used widely in lithium-ion batteries because of its good ionic conductivity in LIB and its mechanistically pathways described by Meyer et al. [17]. Oligo (Ethylene Glycol) (OEG) is a hydrophilic molecule that has been widely used as a side chain in conjugated polymers. Compared to alkyl chains, OEG chains possess characteristics such as hydrophilicity, high polarity, high flexibility, and ionic conductivity [18]. These properties greatly affect the opto-electronic properties, molecular stacking behaviors in thin films, and opto-electronic device performance of the resulting conjugated polymers. Conjugated polymers bearing OEG side chains can not only show improved opto-electronic device performance but also enable new opto-electronic device applications. Additionally, OEG side chains enable the polymers to dissolve and be processed in environmentally friendly and costeffective non chlorinated solvents, such as water [19].
Figure 1:Chemical Structures of designed PEG N-substituted pyrrole derivatives as building blocks for (left) and their use in the polymerized form in potential applications with function-alized electrodes (right).

We introduce the synthesis pathways to design ethylene glycol derivatives with pyrrole functionality to enable electrochemical depositing on electrode surfaces with additional ionic and electronic functional properties of its deposited film. We describe a way to successively extend N-ethylene glycol pyrrole derivatives by Tri ethylene groups using tetrahydropyranyl THP Protecting Group (PG) chemistry and the corresponding building block that enables the direct N ethylene glycolation of pyrrole and or extents the PEG tale OH Group by another TEG group. Further, we demonstrate a synthetic route of an analogous bipyrrole that is used as a preformed pseudo-crownether in conjunction with suitable ions and lastly, we show a synthetic route of a crown ether pyrrole derivative that can be isolated in high yields. Our design concept is based, for example, on the functional units ethylene glycols used in LIB solid electrolytes, which should ideally be present aprotically in a LIB battery due to the cell chemistry, and an electrolyzable function, pyrroles.
Result and Discussion
Retrosynthesis and synthesis planning
Figure 2 left shows the retrosynthesis steps of oligoethylene glycol-modified pyrroles with the aim of the synthesis of linear oligoethylene glycols. Access to the hexaethylene glycol-modified pyrrole 8 is to be realized by a Williamson ether synthesis from the triethylene glycol-modified pyrrole 6 obtained by N-alkylation. The electrophiles 2 and 4 required for the N-alkylation of pyrrole 5 are to be built up starting from triethylene glycol 1. The key step here is the monotosylation of symmetric alcohols [20]. From the obtained tosylates, access to the corresponding bromides can then be obtained by means of Finkelstein exchange [21].
Figure 2:Retrosynthesis route of linear ethylene glycol pyrrole (left) and cyclic crown ether-modified bipyrrole (right).

Crown ether 15 was previously synthesized by Peters et al. [22], but their synthetic route was associated with very low yields (<5%) and could not be reproduced in previous in-house synthesis attempts. Therefore, a different strategy was followed to construct crown ether 15, which is shown in (right). This was prepared following previously described methods for the synthesis of crown ethers [20,23,24]. The crown ether-modified bipyrrole 15 is to be prepared from the monotosylated bipyrrole 18 by intramolecular ring closure reaction, in highly dilute solutions. Key step here is again the monotosylation of symmetric alcohols, which will be used to verify the access to the monotosylated bipyrrole 18. The incorporation of the oligoeth-ylene glycol groups is to be achieved by N-alkylation of bipyrrole 10. The preparation of elec-trophiles 2 and 4 is to be carried out in the same manner as for the linear oligoethylene glycol-modified pyrroles. The bipyrrole 10 can be accessed either by oxidative coupling [25,26] or by Vilsmeier reaction [27,28] from pyrrole 5 and pyrrolidin-2-one 17. The product of the Vilsmeier reaction 9 is commercially available, and therefore this synthesis step was not considered. The product 9 of the Vilsmeier reaction is to be converted into the bipyrrole 10 by means of oxidation with Pd/C [27].
In the following, we describe the successfully performed synthetic routes leading to the oligoethylene glycol-pyrrole derivatives and bipyrrole derivatives 6, 8, 12 and 13, respectively. All new products were proofed and characterized by means of 1Hund 13C-NMR, FTIR and TLC Refraction index (Rf) in the case of column chromatic purification. Intermediates were at least checked by 1H-NMR after isolation if previously described in the literature. Full description and analytical data is described and listed in the experimental part. In the descriptive discussion, we mainly address the yields as the endpoint.
Synthesis of the linear N-Pyrrol-PEG-derivatives
Figure 3 shows the four synthesis steps for the target compound PyTEG-PG (monomer 6) shown at the beginning. The intermediate and latter building block 4 is directly reacted with the pyrrole 5. Deprotection of the Protecting Group (PG) of 6 and renewed reaction with 4 leads to Py(TEG)2-PG (monomer 8) by the analogue of 6 extended by a TEG group. Repetitive synthesis steps of a deprotection group and reaction with 4 thus led to the linear N-pyrrole derivatives of the form Py(TEG)n+1-PG. This allows a successive extension of the ethylene glycol moiety in the functionalized N-alkylated pyrrole derivative by three (CH2-CH2-O)3 units.
Figure 3:Synthesis steps and condition of building blocks 4, 6 and 8 that allows successive buildup of linear designed pyrrole-ethylenglycol derivatives of the structure Py(TEG)n+1-PG for latter electrolpolymerisation on electrodes.

The starting substance is the Triethylene Glycol (TEG). For monotosylation, we followed a synthetic route of Bouzide et al. [20] which describes a method for selective monotosylation, which does not require an excess of diol and with mild reaction conditions, while maintaining high yields of 85% of the tosylated Tos-TEG-OH (2), whose free OH group could be synthe-sized with protecting group chemistry to give monomer 4 and also isolated with high yields of 92%. In the following, the pyrrole, which anionically substitutes the bromide from 4 under strongly alkaline conditions and converts to 6 with yielding 70%.
Synthesis of oligo ethylene glycol functionalized bipyrroles
Figure 4 shows the synthesis route of the N,N-BiPy-(TEGPG) 2(12) that formally corresponds to the dimer of N-Py-TEG-PG 6. We additionally designed and synthesized this compound after encountering non-sufficient electro polymerizable properties with the produced N-pyrrole derivative 13 modified with a crown ether as residue. N,N-BiPy-(TEG-OH)2 is known to build up in the presents of Li+ ions to change its structure to a pseudo-crown ether [29]. Secondly, we did not succeed in reproducing the in the literature described in the procedure with already very low yields described of approx. 2-5% for the crown ether modified bipyrrole in both of ours inhouse labs. However, the shown synthesis of 12 is, for both of these aspects we encountered, a worthwhile method to obtain an isolable substance that is feasible present-able in the laboratory and is available as an alternative for a polymerizable and with a pseu-do-crown ether precursor formable compound.
Figure 4:Synthesis route of 1,1’-bis(2-(2-(2-((tetrahydro-2H-pyran-2-yl)oxy)ethoxy)ethoxy)ethyl)-1H,1’H-2,2’- bipyrrole.

Starting from 2,2’-(pyridinyl)-pyrrole 10. This was converted to the 2,2’-bipyrrole by oxidation with Pd/C, with a yield of 29.5% according to the described method by Geier et al. [27] and extended refurbishing procedure from Kauffmann et al. [30] which was due to the great sensitivity of the compound to light, air and temperature [28,30]. The next step consisted of the alkylation of the 2,2’-bipyrrole with the above-described building block 4 yielding 23%. Also the further synthesis of 12 by deprotecting the compound and then subjected to Wil-liamson ether synthesis conditions as previously described by Peters et al. [22] with the exact same compounds was yielded zero percentage and let us follow another path, which will be described next.
Synthesis of the benzo-crownether modified pyrrole
Figure 5:One step synthesis of 1-(2,3,5,6,8,9,11,12,14,15-decahydrobenzo[b][1,4,7,10,13,16] hexaoxacyclooctadecin-18-yl)-1H-pyrrole 13.

Former synthesis description of crownether modified pyrrole derivatives are based on multistep synthesis steps in which ring closure of the cyclic ether must take place in the last step, and this goes along with very low yields in the low single-digit percent range. This is true for the pyrrole derivative as well as for the bipyrrole attempts. Since we did not succeed to verify the above description, we switched to a direct the Clauson-Kaas type reaction [31] with 2,5-Dimethoxytetrahydrofuran 14 and crownetherbenzoanilin 12 (Figure 5) - according to the reaction condition Azizi et al. [32] described-to the aimed compound 14 yielding 40% only and observing a high content of black polymerized raw product which is obviously due to a premature chemical polymerization of the pyrrole derivative.
Electrochemical polymerization of the monomers
Electrochemical polymerization was performed potentiodynamically using Cyclic Volt-ammetry (CV) to obtain information about the growth of the polymer layer and to determine the polymerization potential. The onset of polymerization is evident from the characteristic irreversible peak, during the first scan (Figure 6). This differs from the subsequent cycles and represents the nucleation of the polymer at the metal surface [33,34] or this peak is also referred to as the “nucleation loop” [35].
Figure 6:Electro polymerization scheme and its electrochemical stoichiometry of 6,7,8 (above) and 12 (below). Inlet photo shows the black deposit of the polypyrrole-derivatives on a blanc Pt-disc-electrode.

After determination of the electro polymerization potential by successively increasing the polymerization voltage and then evaluating the Qc/Qa quotient of the reversible polypyrrole redox system formed (see methods part) it allows a comparative evaluation of the electro-chemically deposited polypyrrole layers. If the ratio Qc/Qa corresponds <1, either irreversible overoxidation of the polymer sets in or it indicates the formation of soluble oligomers, which are not involved in the polymer growth [36,37]. The optimum polymerization potential, while maintaining the reversible redox system, was determined between 1.0-1.2 V vs. Ag/ AgCl for all monomers.
For all CVs shown (Figure 7) in each new cycle, the current increases in the range of 0.8V due to the formation of the polypyrrole film, which has its own reversible redox system. This correlation reflects the increase in the surface area of the polymer as well as the increase in the number of chargeable redox sites in accordance with the detailed studies of Heinze et al. [33]. After the monomers are oxidized in the anodic region, their deposition is followed by cathodic reduction. In the following cycle, the deposited polymer is oxidized again, and the redox system of the polymer is formed. From the cyclic voltammograms, the peak potential EpaPol of the resulting polymer is obtained, these are summarized as EpaPol for each monomer in Fehler! Verweisquelle konnte nicht gefunden werden. The polymerization potentials Epa are similar for monomers 6, 7 and 8 and are in the typical range for N-alkylated pyrroles at a Pt electrode. Also, the peak potentials EpaPol of the resulting polymer are, for all monomers, approximately in the same voltage range of 0.81V - 0.88V and correspond to the reported redox potentials of polypyrrole [38,39]. It is noticeable that as the number of ethylene glycol units increases, the peak potential of the polymers EpaPol increases. The cyclic voltammograms of the THP-protected oligoethylene glycol-modified pyrroles 7 and 8, show a significantly lower growth of the redox system. The reason is explained by the electrically insulating properties of the protecting group. The oligoethylene glycol units also lack electron conducting properties, but the influence here does not appear to be so dominant. The steric influence of the pyran ring could also have a reducing effect by separating the conductive polypyrrole layers on the electrochemical deposition.
Figure 7:Cyclic voltammograms of electrochemical polymerization of the monomer 9 (a), 10 (b), 8 (c) and 6 (d) from 0.025 M monomer solution in 0.1 M TBABF4/MeCN, with 25 cycles (every 5th is plotted) for each polymerization, scan rate v=100mV/s. Potential [V] vs. Ag/AgCl.

The electrochemical polymerization of monomers 7 and 12 theoretically yields the same polymer, although monomer 12, due to its bipyrrole structure, requires only half the number of electrons to arrive at the same polymer chain length. The structural difference between the two monomers is noticeable in the polymerization behaviour. It can be seen clearly in Figure 7 and Table 1 Fehler! Verweisquelle konnte nicht gefunden werden. that the polymerization potential and thus the oxidation potential of 2,2’-bipyrrole 12 is 200mV lower than that of the other monomers. The cyclic voltammogram of 2,2’-bipyrrole 12, further shows a shoulder at 0.8V which has been observed before by Rohde et al. [40] in the polymerization of oligo-(1-methylpyrroles) and was attributed to conformational changes [41].
Table 1:Summarized results from the analysis of the electrochemically deposited monomers 6,7,8,12 (EpaMon) and their corresponding polypyrroles P1-P4 obtained after. Q values were ob-tained from the CV results while polymerization.

Amount and mass of deposit monomers
The monomer mass deposited was determined as described in section method. The charge Q was determined from the integrals of the peak areas in the 25th cycle. Table 2 shows the deposited amounts of the theoretical material and masses, as well as the amount of material of the electrons, after 25 cycles, neglecting doing processes in the polymer films.
Table 2:Deposited amounts of material and masses as well as amount of material of electrons for each polymerization.

The direct comparison of the deposited number of monomers 7 and 6, which differ structurally only in the protective group, shows the negative influence of the THP end group on the polymerization. Under the same polymerization conditions, significantly fewer monomers are deposited from the protected monomer 7 than from the unprotected monomer 6. The reasons for this might be due to a) the electrically insulating properties of the THP end group, b) to the steric effect of the pyran ring, which causes a reduced electrical conductivity or c) represent the solubility of the resulting oligopyrrole derivative in the electrolyte solution, so that polymer products are detached from the surface by solution processes and whose share is eluded by the method used.
Thus, it can be assumed that higher deposition rates can be achieved using the sterically less demanding methyl ether end group, rather than the THP end group. More monomers are deposited from the also protected 2,2’-bipyrrole 12, in contrast to the protected monomer 7. This relationship is due to the increased reactivity of the 2,2’-bipyrroles, which is also evident in the kinetics of this monomer [33]. Further advantageous about the 2,2’-bipyrrole 12 is that two pyrrole units are already deposited per monomer unit. Thus, longer polymer chains are accessible with the bipyrrole 12 as monomer. Figure 8 shows the increase of electro-deposited polymer vs. number of CV cycles, which is strictly linearly correlated for all species and shows that the concentration decreases near the electrode while the polymerization has no decisive influence under the chosen condition.
Figure 8:Amount of monomer deposited as a function of the number of cycles.

Over-oxidation and impedance spectroscopy
The electrical conductivity of the deposited polypyrrole films is counterproductive in the intended functionality of the deposited polypyrrole layers as a solid electrolyte in e.g. an energy storage system. Therefore, the deposited polypyrrole derivatives were irreversibly overoxidized on the surface at high voltages in a second process step, which destroys the conjugated double bonds of the polypyrrole backbone chains (Figure 9 left). To determine the overoxidation potential, the polymer films were transferred to electrolyte solution and the voltage was increased in 0.1V steps with each new measurement. It was found that irreversible overoxidation starts at a potential of 1.9V. The polymers then exhibit a high overoxidation potential. The polymers no longer exhibit any electrochemical activity in the subsequent scan at 2V. The CVs in Figure 9 show that at potential of 1.3V (blue cyclic voltammogram) the redox system and thus the electron conductivity of the polymer film is completely preserved. After overoxidation at 1.9V (green cyclic voltammogram), only the background noise in the range of the previous redox system can be recognized. Thus, the polymer films no longer exhibit elec-trochemical activity after overoxidation, from which it can be concluded that there is no longer any electrical conductivity. After the overoxidized polymer films were dried, they changed color from black to yellowish, golden color.
Figure 9:CV of polymerfilm P3 before (black) and stepwise overoxidation (red, blue and green) in monomer free electrolyte; photos of P3 covered Pt-disk electrode (diameter of 3mm) before and after overoxidation (right).

The ionic conductivities of the dried and overoxidized polymer films were determined by impedance spectroscopy (Figure 10). The polymer films were previously wetted with 0.1 M LiClO4 in MeCN in order to feed the polymer film with lithium salt and put in a battery cell against stainless steel for measurements. The calculated lithium-ion conductivities are shown in Table 3. Furthermore, it was not possible to determine the film thickness of the polymer films directly and we are calculating this parameter from The EIS fit assuming a capacitor. The area of the electrode was 0.07cm². The polymer film of P4 came off immediately after treatment and evaded further EIS examination.
Figure 10:Nyquist plot (above) and bode plot (middle) of P1 with Kramers-Kronig Fit (blue line), double CPE Fit (red line) and fit according to Iroh et. al; circuit models (below left: CPE based model and below right model according to Iroh).

Table 3:Measured resistivities and calculated ionic lithium conductivities of polymers P1-P3. κ= d/(Rct*A) with d=layer thickness, A=surface of electrodes. *Calculating from the EIS capacity for P1 assuming a plate condensator. The thickness error is +/- 16% due to the uncertainty of εr.

**Calculating a linear dependence on different mass relative to P1.
d=(ε₀*εr*A)/C (1)
Where ε₀ is the electric field constant (8.854x10-12F/m), εr=12+/-2 (the relative dielectric constant of OPPy taken from [42], A is the area (0.07cm²) of the used electrodes, and C (see Cc value in Fehler! Verweisquelle konnte nicht gefunden werden. 4) is the measured capacitance from the fitted EIS model.
Materials and Methods
General electrochemical measurements
All electrochemical experiments were performed with a Ptelectrode (Kel-F body 932-03, Gamry USA) of surface 0.07cm² as a working electrode. The counter electrode also consisted of Pt (A=0.15cm2), and a 12cm reference electrode from Metrom (Germany) was used.
Electro polymerization of the monomers
The electrochemical polymerization of all monomers was achieved in a 0.025M monomer solution in 10mL 0.1M TBABF4 in MeCN. Before each polymerization, the solution was rinsed with argon for 10min. The determination of the optimal polymerization potential was determined by increasing the voltage from a potential of 0.9V by 0.05V at each measurement. The Pt electrode was cleaned before each new measurement. With each polymerization, 25 cycles were completed (Table 4).
Table 4:Obtained parameter results of the model according to Iroh (green line) from P1. N=3.

Before, the polymerization potentials were determined by increasing the voltage at the working electrode stepwise by 0.05V for each new measurement. To ensure that each measurement was made under the same conditions, the Pt electrode was cleaned with each new measurement. At the appearance of the first characteristic peak, 25 cycles were performed at this potential. This showed that polymerization at this potential resulted in a reversible redox system of the polymer. Experiments on polymerization at higher potentials showed an increase in the irreversibility of the redox system which was expressed in the decrease in the cathodic peak currents ipc and in the ratio of the peak areas Qc/Qa (charge of both processes). The cathodic charge Qc and the anodic charge Qa were calculated by integrating the peak areas after 25 cycles in each case.
Calculation of the deposited monomer mass and amount of substance: The Pt electrode with the generated polymer film was washed several times with electrolyte solution before it was transferred to an electrochemical cell containing 20mL of 0.1M TBABF4/MeCN. Again, the electrolyte solution was flushed with argon for 10min before each measurement. The polymer films were measured at different scan rates ranging from 100mV/s - 1000mV/s in 100mV/s steps and ranging from 1000mV/s - 2000mV/s in 250mV/s steps. One cycle was run at each scanning rate:
(2MU+4H) (n+1)=(nMU+2H+2MU)(nx)+ + (2n+2)H+ + (2n+2+nx)e- (1)
With MU=Monomer Unit=MMonomer-2H, n=amount of monomers in mol, x=partial oxidation of the Polymer with an assumed value of 0.25F/mol.
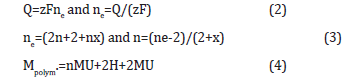
Mpolym.=mass of deposited Polymer; With z=charge number of the Ion, Q=electric charge, F=Faraday constant=96485 C/mol, ne=amount of electrons
Experimental section
NMR-spectra were recorded with a Unity INOVA 500 NB (Varian, USA). The FT-IR spectra were recorded with a Scimitar 2000 (Varian, USA). All air moisture sensitive reactions were carried out under Ar2 using oven-dried glassware. 2-[2-(2-hydroxyethoxy) ethyl]-4-methylbenzenesulfonate (2)
A stirred solution of 4.46mL (33.3mmol) 1 triethylene glycol in anhydrous CH2Cl2 (230mL ) was cooled with ice to 5 °C. Then 11.58g (49.95mmol) Silver-(I)-Oxide, 6.98g (36.63mmol) p-toluenesulfonyl chloride and 1.11g (6.66mmol) potassium iodide were added. The reaction mixture was stirred at 5 °C for 20min, then filtered through a small pad of silica gel, and washed with EtOAc. Evaporation of the solvent, followed by column chromatography (SiO2/EtOAc) gave 8.61g (28.31mmol, 85%) of the desired monotosylate ester 2. 1H-NMR (500MHz, CDCl3, TMS): δ (ppm)=7.80 (d, J=8.2Hz, CHarom, 2H), 7.32 (d, J=8.2Hz, CHarom, 2H); 4.18 – 4.13 (m, CH2-OTs, 2H), 3.70 (q, J=4.4, 4.2, 4.2Hz, CH2-O, 4H), 3.60 (s, CH2-O, 4H), 3.58 – 3.55 (m, CH2-OH, 2H), 2.43 (s, CH3, 3H). FT-IR (ATR): ν̃ (cm-1)=3453b, 2936s, 2874s; 1352s, 1097s, 915s, 751 s, 660 s, 549 s. TLC: Rf=0.40 (EtOAc)
2-(2-(2-((tetrahydro-2H-pyran-2-yl)oxy)ethoxy)ethoxy) ethyl-4-methylbenzenesulfonate (3). To a stirred solution of 17.86g (58.73mmol) TsO-OEG3-OH 2 and 7.96mL (117mmol) 3,4-Dihydropyran in CH2Cl2 (411mL ) was added 2.95g (11.75mmol) Pyridinium p-toluenesulfonate, and the reaction mixture was stirred at r.t. for 2h. TLC monitored the progress of the reaction. When the TsO-OEG3-OH 2 was no longer present, the reaction mixture was transferred to a separating funnel and diluted with EtOAc (400mL). The solution was washed with saturated NaCl solution (3x200mL), and the aqueous layer was extracted with EtOAc (3x100mL). The organic layers were combined and dried over MgSO4. After the filtration, all volatiles were evaporated in vacuo to obtain 21.34g (54.93mmol, 94 %) of the protected tosylate 3 which was used without further purification in the following step.1H-NMR (500MHz, DMSO-d6, TMS): δ (ppm)=7.80 (d, J=8.2Hz, CHarom, 2H), 7.49 (d, J=8.2Hz, CHarom, 2H), 4.57 (m, O-CH-ORTHP, 1H), 4.11 (m, CH2-OTs, 2H), 3.76 (m, CH2-OTHP, 2H), 3.71 (m, CH2-OTHP, 1H), 3.59–3.46 (m, CH2-O, 10H), 3.42 (m, CH2-OTHP, 1H), 2.42 (s, CH3, 3H), 1.73–1.44 (m, CH2-THP, 6H). FT-IR (ATR): ν̃ (cm-1)=2938s, 2869s, 1597m, 1450m, 1289s, 1354s, 1175s, 1121s, 1022s, 915s, 811s, 769s, 661s. TLC: Rf = 0.77 (EtOAc).
2-{2-[2-(2-bromoethoxy)ethoxy]ethoxy}tetrahydro-2H-pyran (4). To a stirred solution of 21g (54.05mmol) TsO-OEG3 -OTHP 3 in acetone (260mL) was added 7.04g (81.08mmol) LiBr under argon atmosphere. After a few minutes, the reaction mixture became cloudy due to the precipitation of the LiOTs. The reaction mixture was stirred for 7h at r.t. and then transferred to a separating funnel. Then H2O (200mL) and Et2O (300mL) was added, and the layers were separated. The organic layer was washed with H2O (3x100mL), and the aqueous layer was extracted with EtO2 (2x100mL). The organic layers were combined and dried over MgSO4. After the filtration, the solvent was removed under reduced pressure, and the transparent, liquid residue was purified by column chromatography (SiO2, EtOAc/n-hexane 2:1). 10.02g (37.05mmol, 67%) of the protected bromide 4 was obtained as a colorless liquid.1H-NMR (500MHz, CDCl3, TMS): δ (ppm)=4.62 (m, O-CH-ORTHP, 1H), 3.85 (dt, J=10.6, 4.4Hz, Br-CH2-CH2, CHH-OTHP, 3H), 3.80 (t, J=6.3, 6.3 Hz, CH2-OTHP, 2H), 3.67(s, CH2-O, 6H), 3.59 (dt, J=10.6, 4.9, CHH-OTHP, 1H), 3.47 (t, J=6.0, 6.0Hz, CH2-Br, 2H), 1.84–1.49 (m, CH2-THP, 6H). FT-IR (ATR): ν̃ (cm-1)=2936s, 2867s, 1449m, 1351m, 1279m, 1117s, 1029s, 985s, 971s, 812m, 663m. TLC: Rf=0.75 (EtOAc/n-heptane 2:1).
1-(2-(2-(2-((tetrahydro-2H-pyran-2-yl)oxy)ethoxy)ethoxy) ethyl)-1H-pyrrole (6). An oven-dried flask was charged with DMSO (15mL) followed by the addition of 1.75g (15.65mmol) KOtBu and 0.32g (1.2mmol) 18-Crown-6. After 10min of stirring 830μL (12.02mmol), fresh distilled pyrrole 5 in DMSO (10mL) was added dropwise for 20min. The reaction mixture was warmed to 65 °C and keep there for 1.5h. The yellow reaction mixture was then cooled to 0 °C followed by the addition of 5g (16.83mmol) of the protected bromide 4 in DMSO (15mL) dropwise over 20min. The ensuing mixture was headed to 65 °C, stirred at this temperature for 18h then transferred to a separating funnel. Than H2O (100mL) and EtO2 (100mL) was added, and the resulting layers were separated. The organic layer was washed with H2O (3x100mL), and the aqueous layer was extracted with Et2O (3x100mL). The combined organic extracts were dried over MgSO4, filtered and concentrated under reduced pressure to give a yellow oil. The subjection of this oil to column chromatography (SiO2, EtOAc/n-Heptane 2:1) gave 2.39g (8.42mmol, 70%) of the protected and N-alkylated pyrrole 6 as a yellow oil.
1H-NMR (500MHz, CDCl3, TMS): δ (ppm)=6.71 (s, N-CHPyrrol, 2H), 6.14 (s, CHPyrrol, 2H), 4.62 (m, O-CH-ORTHP, 1H), 4.07 (t, J=5.5Hz, 5.5Hz, N-CH2, 2H), 3.87 (m, CH2-OTHP, 2H), 3.75 (t, J=5.5Hz, 5.5Hz, N-CH2CH2, 2H), 3.72 (m, CH2-OTHP, 1H), 3.64–3.56 (m, CH2-O, 6H), 3.51 (m, CH2-OTHP, 1H), 3.49 (m, CH2-OTHP, 1H), 1.84–1.49 (m, CH2-THP, 6H) FT-IR (ATR): ν̃ (cm-1)=3102w, 2934s, 2867s, 1542m, 1498m, 1445m, 1351m, 1284m, 1201m, 1119s, 1073s, 1029m, 984s, 929w, 871w, 813w, 722s, 615m, TLC: Rf=0.35 (EtOAc). 2-[2-(1H-Pyrrol-1-yl)-ethoxy]-ethan-1-ol (7). A stirred solution of 3.99g (14.1mmol) the protected and N-alkylated pyrrole 6 in EtOH (125mL) was headed to 55 °C. Then 375mg (1.41mmol) Pyridinium p-toluenesulfonate was added, followed by stirring for three hours at 55 °C. Next, the reaction mixture was cooled to r.t., extracted with EtOAc and concentrated at reduced pressure. The residue was purified by column chromatography (SiO2, EtOAc/n- Heptane 2:1) affording 2.6g (13.11mmol, 93%) of the pyrrole 7 as a yellow liquid. 1H-NMR (500MHz, CDCl3, TMS): δ (ppm) = 6.71 (s, N-CHPyrrol, 2H), 6.14 (s, CHPyrrol, 2H), 4.07 (t, J=5.5Hz, 5.5Hz, N-CH2-CH2, 2H), 3.75 (t, J=5.5Hz, 5.5Hz, N-CH2, 2H), 3.73-3.71 (m, CH2-O, 2H), 3.65- 3.63 (m, CH2-O, 2H), 3.59–3.57 (m, CH2-O, 4H) FT-IR (ATR): ν̃ (cm-1)=3437b, 3102w, 2868m, 1733s, 1499s, 1446s, 1354m, 1284m, 1244m, 1116s, 1063s, 925m, 886m, 723s, 654m, 615m, 558w Rf=0.25 (EtOAc)
1-{17-[(tetrahydro-2H-pyran-2-yl)oxy]-3,6,9,12,15- pentaoxaheptadecyl}-1H-pyrrole (8). A suspension of 157mg (6.54mmol) NaH in DMSO (10mL) was prepared in a threeneck round-bottom flask, and the suspension was cooled to 0 °C, followed by the addition of 1g (5.03mmol) pyrrole 7 in DMSO (5mL). The reaction mixture was allowed to reach r.t. and then stirred for 1.5h at this temperature. The reaction mixture was subsequently re-cooled to 0 °C, followed by the addition of 1.64g (5.53mmol) the bromide 4 in DMSO (5mL) dropwise for 20min. The resulting mixture was allowed to reach r.t. and kept there for 18h. The reaction was quenched with MeOH (15mL), and the resulting NaOMe was removed via a frit. The solution was washed with EtOAc and concentrated at reduced pressure. The yellow, oily residue was purified by column chromatography (SiO2, EtOAc/n- Hexane 2:1) to obtain 600mg (1.45mmol, 29%) of the pyrrole 8 as a yellow liquid. 1H-NMR (500MHz, CDCl3, TMS): δ (ppm)=6.71 (s, N-CHPyrrol, 2H), 6.14 (s, CHPyrrol, 2H), 4.62 (m, O-CH-OR-THP, 1H), 4.07 (t, J=5.5Hz, 5.5Hz, N-CH2, 2H), 3.87 (m, CH2-OTHP, 2H), 3.75 (t, J=5.5 Hz, 5.5Hz, N-CH2CH2, 2H), 3.72 (m, CH2-OTHP, 1H), 3.66–3.56 (m, CH2-O, 20H), 3.49 (m, CH2-OTHP, 1H), 1.84 – 1.49 (m, CH2-THP, 6H). 13C-NMR (126MHz, CDCl3, TMS): δ (ppm)=120.9 (N-CHPyrrol), 108.0 (CHPyrrol), 98.9 (O-CHR-OTHP ), 70.5 (CH2-O), 69.4 (N-CH2-CH2), 65.2 (CH2-OTHP), 62.2 (O-CH2-THP ), 49.4 (NCH 2), 30.5, 25.4, 19.6 (CH2-THP). MS-ESI (+): m/z (%)=433.4 (100) [M+NH4]+, 416.5 (32) [M+H]+, 398.5 (90) [M -NH4]+. FT-IR (ATR): ν̃ (cm-1)=3100w, 2928s, 2867s,1736m, 1497m, 1445m, 1351m, 1285m, 1245m, 1201m, 1121w, 1112s, 1029s, 986s, 935w, 870m, 813m, 724s, 615m, Rf=0.15 (EtOAc).
4’,5’-dihydro-1H,3’H-2,2’-bipyrrole (9). To a solution of 60.3g (899mmol) of pyrrole 5 in CH2Cl2 (40mL), 28.2g (184mmol) of POCl3 are slowly added under ice cooling. To this mixture 17.5g (205.6mmol) of pyrroli-din-2-one are added very slowly. The reaction mixture is then stirred for a further 3h without cooling. To isolate the target product, first dilute with CH2Cl2 (100mL) and add the completely homogeneous solution in portions to a concentrated NaOH solution under ice cooling. After this hydrolysis, the pH value is further raised to pH10 by adding 10M NaOH (ca. 32%, approx. 100g). After phase separation, extraction is carried out twice or more with CH2Cl2 (80ml). The evaporation of these extracts would yield a lot of polymer. Therefore, the organic extract is extracted several times with 0.5M HCl (800g) and the target product is precipitated as a terra-cotta-coloured solid by adding 5M NaOH (approx. 80g) drop by drop. The raw product is re-solved again in CH2Cl2 (250mL) and dried with NaSO4. After separation of the solvent 23.8g raw product is obtained, which is purified by sublimation at 160° in vacuum. This leaves 11.8g (43%; mp. 163 °C) of the target product 9 as white crystalline solid.
1´H, 1H - 2, 2´-Bipyrrole (10). A stirred solution of 2.01g (15mmol) 10 in diglyme (150mL) under Argon atmosphere was heated to 70 °C to dissolve 10. To the solution was added 2.01g (1.97mmol Pd) 10% palladium on carbon, the reaction mixture was heated to reflux and kept there of 1.5h. After cooling to r.t. the Pd/C was passed through a fluted filter, and the collected palladium on carbon was washed with DCM. The solution was filtered through a small pad of celite, washed with H2O and dried over MgSO4. The ensuring green solution was concentrated at reduced pressure to afford a grey Residue. The crude 11 was purified by sublimation (100 °C, 5.4*10-4mbar) to obtain 586mg (4.43mmol, 29.5%) 11 as a with crystalline solid. 1H-NMR (500MHz, CDCl3, TMS): δ (ppm)=8.29 (br s, NH, 2H), 6.87 (s, N-CHPyrrol, 2H), 6.20-6.25 (m, CHPyrrol, 4H). TLC: Rf=0.37 (Toluene/DCM 3:1).
1,1’-bis(2-(2-(2-((tetrahydro-2H-pyran-2-yl)oxy)ethoxy) ethoxy)ethyl)-1H,1’H-2,2’-bipyrrole (11). A stirred solution of 355mg (8.86mmol) KH and 114mg (0.88mmol) 18-Crown-6 in DMSO (10mL) under Argon atmosphere was cooled to 0 °C. After 10min of stirring, 586mg (4.43mmol) Bipyrrole 11 in DMSO (5mL) was added dropwise over 20min. The resulting green reaction mixture was allowed to reach r.t. and was stirred for 1.5h. After re-cooling to 0 °C 2.89g (9.75mmol) 4 in DMSO (5mL) was added dropwise for 20min. The reaction mixture was allowed to reach r.t. and was stirred for 48h at this temperature. Than tert-Butanol (10mL) was added to quench the reaction. Next, the mixture was transferred into a separation funnel, and the layers were separated. The organic layer was washed with H2O (3x75mL), and the aqueous layer was washed with EtOAc (3x50mL). The combined organic phases were dried over MgSO4, and the MgSO4 was removed with a folded filter. The filtrate was removed from the solvent under reduced pressure. The ensuring dark red residue was directly subjected to column chromatography (SiO2, EtOAc/n-Heptane) to obtain 570mg (1.01mmol, 23%) of the alkylated Bipyrrole 12 as a dark red oil. 1H-NMR (500MHz, CDCl3, TMS): δ (ppm)=6.87 (s, N-CHPyrrol, 2H), 6.17-6.14 (m, CHPyrrol, 4H), 4.63 (s, O-CH-OR-THP, 2H), 3.94-3.92 (m, N-CH2, 4H), 3.86-3.84 (m, N-CH2-CH2, 4H), 3.71 3.49 (m, CH2-O, CH2- OTHP, 20H), 1.86–1.53 (m, CH2-THP, 12H) 13C-NMR(126MHz, CDCl3, TMS): δ (ppm)=124.1 (C-C-Bipyrrol), 122.1 (N-CHPyrrol), 111.2, 107.5 (CHPyrrol), 98.9 (O-CHR-OTHP), 71.2 (CH2-O), 70.6 (N-CH2CH2), 66.6 (CH2-CH2-OTHP), 62.2 (CH2-OTHP), 46.4 (N-CH2), 30.5, 25.4, 19.5 (CH2-THP). MS-ESI (+): m/z (%) = 582.3 (100) [M+NH4]+, 565.4 (86) [M+H]+, 546.4 (33) [M-NH4]+. FT-IR (ATR): ν̃ (cm- 1)=3101w, 2935s, 2867s, 1445m, 1351m, 1284m, 01120s, 1074s, 1030s, 871w, 814w, 723s, 618w TLC: Rf=0.36 (EtOAc/n-Heptane 2:1) 1-(2,3,5,6,8,9,11,12,14,15-decahydrobenzo[b][1,4,7,10,13,16] hexaoxacyclooctadecin-18yl)-1H-pyrrole (13).
A stirred solution of 500mg (1.53mmol) 4’-Aminobenzo-18- Crown-6 12 and 258μL (2mmol) 2,5-Dimethoxytetrahydrofuran in H2O (3mL) was heated to 60 °C. After the reaction mixture had reached a temperature of 60 °C, 12.4mg (46μmol) FeCl3*6H2O was added, and the reaction mixture turned dark red. The reaction mixture was stirred for 4h at 60 °C. After the reaction mixture had cooled down to r.t, it was extracted with EtOAc (60mL). The FeCl3*6H2O was removed with a G-4 frit, and the Filtrate washed with EtOAc. The solvent was removed under reduced pressure, after which a black, oily residue remained. The Subjection of this black oily residue to column chromatography (SiO2, EtOH/nheptane 4:1) gave 222mg (0.59mmol, 39.9%) of the benzo-crownether- modified Pyrrole 13 as a colourless solid. The blackish residue remained inside the column. 1H-NMR (500MHz, DMSO-d6, TMS): δ (ppm)=7.28 (s, N-CHPyrrol, 2H), 7.13 (m, CHBenzol, 1H), 7.02 (m, CHBenzol, 2H), 6.25 (s, CHPyrrol, 2H), 4.19 (m, Car-om-O-CH2, 2H), 4.10 (m, Carom-O-CH2, 2H), 3.77-3.56 (m, O-CH2, 16H) 13C-NMR (126MHz, DMSO-d6, TMS): δ (ppm)=149.16 (Carom-O-R), 146.25 (Carom-O-R), 143.37 (N-CHarom), 119.74 (N-CHPyrrol), 113.96 (Carom), 111.74 (Carom), 110.25 (CHPyrrol), 70.20 (O-CH2), 69.14 (Carom-O-CH2) MS-ESI (+): m/z (%)=400.4 (20) [M+Na]+, 395.4 (100) [M+NH4]+, 378.4 (27) [M+H]+, Rf=0.41 (EtOH/n-Heptane 4:1).
Conclusion
This study presented the synthesis and characterization of four N-substituted pyrrole derivatives functionalized with oligo(ethylene glycol). Electrodeposition of these derivatives from acetonitrile electrolytes onto a platinum electrode was successfully demonstrated using cyclic voltammetry. Three stable deposited films were characterized based on their electro polymerizable behavior, and the deposited masses were calculated using the evaluated charges of the reversible redox peaks of the polypyrrole systems.
The overoxidation of the polypyrrole redox system potentials was determined, and impedance spectroscopy was employed to assess Li+ ion conductivity within the deposited film P1. These novel compounds hold potential contributions to the field of electrode modification with polypyrrole, which finds applications in electronics, batteries, supercapacitors, and sensors.
Author Contributions
W.M. acquired the funding, managed the project, and wrote the manuscript; P.S. planned and performed the experiments, analyzed the data, and wrote the manuscript in his master thesis. J.S: performed two synthesis and reviewed the manuscript. All authors have read and agreed to the published version of the manuscript.
Acknowledgment
The financial support within the project Batt3D by the German Federal Ministry of Economic Affairs and Energy (BMWI) is gratefully acknowledged.
References
- Bof Bufon CC, Heinzel T (2006) Polypyrrole thin-film field-effect transistor. Appl Phys Lett 89: 012104.
- Jin B, Gao F, Zhu YF, Lang XY, Han GF, et al. (2016) Facile synthesis of non-graphitizable polypyrrole-derived carbon/carbon nanotubes for lithium-ion batteries. Sci. Rep 6: 19317.
- Yang C, Shen J, Wang C, Fei H, Bao H, et al. (2014) All-solid-state asymmetric supercapacitor based on reduced graphene oxide/carbon nanotube and carbon fiber paper/polypyrrole electrodes. J Mater Chem A 2: 1458-1464.
- Park KS, Schougaard SB, Goodenough JB (2007) Conducting-polymer/iron-redox-couple composite cathodes for lithium secondary batteries. Adv Mater 19(6): 848-851.
- Liu Y, Xu N, Chen W, Wang X, Sun C, et al. (2018) Supercapacitor with high cycling stability through electrochemical deposition of metal–organic frameworks/polypyrrole positive electrode. Dalton Trans 47: 13472-13478.
- Parnell CM, Chhetri BP, Mitchell TB, Watanabe F, Kannarpady G, et al. (2019) Simultaneous electrochemical deposition of cobalt complex and poly(pyrrole) thin films for supercapacitor electrodes. Sci Rep 9(1): 5650.
- Wang J, Too CO, Zhou D, Wallace GG (2005) Novel electrode substrates for rechargeable lithium/polypyrrole batteries. J Power Sources 140(1): 162-167.
- Afzal A, Abuilaiwi FA, Habib A, Awais M, Waje SB, et al. (2017) Polypyrrole/carbon nanotube supercapacitors: Technological advances and challenges. J Power Sources 352(1): 174-186.
- Fan LZ, Maier J (2006) High-performance polypyrrole electrode materials for redox supercapacitors. Electrochem Commun 8(6): 937-940.
- Yadav R, Saini A, Choudhary J, Sardana S, Ohlan A, et al. (2023) High-performance flexible supercapacitor based on morphology tuned polypyrrole/molybdenum disulfide nanocomposites. Energy Storage 5(8): e477.
- Vernitskaya TV, Efimov ON (1997) Polypyrrole: a conducting polymer; its synthesis, properties and applications. Russ Chem Rev 66: 443-57.
- Köhler S (2023) Influence of selected synthesis parameters on the electrochemical and mechanical properties of polypyrrole - German Digital Library.
- Liao B, Hu X, Xu M, Li H, Yu L, et al. (2018) Constructing unique cathode interface by manipulating functional groups of electrolyte additive for graphite/LiNi6CO0.2Mn0.2O2 cells at high voltage. J Phys Chem Lett 9(12): 3434-3445.
- Kim S, Jang LK, Park HS, Lee JY (2016) Electrochemical deposition of conductive and adhesive polypyrrole-dopamine films. Sci Rep 6: 30475.
- Losito I, Malitesta C, Sabbatini L, Zambonin PG (1994) Pristine and overoxidized polypyrrole by XPS. Surf Sci Spectra 3(4): 375-383.
- Shiigi H, Kishimoto M, Yakabe H, Deore B, Nagaoka T (2002) Highly selective molecularly imprinted overoxidized polypyrrole colloids: one-step preparation technique. Anal Sci 18(1): 41-44.
- Meyer WH (1998) Polymer electrolytes for lithium-ion batteries. Adv Mater 10(6): 439-448.
- Meng B, Liu J, Wang L (2020) Oligo (ethylene glycol) as side chains of conjugated polymers for optoelectronic applications. Polym. Chem 11: 1261-1270.
- Chen CA, Wang SC, Tung SH, Su WF (2019) Oligo (ethylene glycol) side chain effect on the physical properties and molecular arrangement of oligothiophene–isoindigo based conjugated polymers. Soft Matter 15(46): 9468-9473.
- Bouzide A, Sauvé G (2002) Silver(I) Oxide mediated highly selective monotosylation of symmetrical diols. Application to the synthesis of polysubstituted cyclic ethers. Org Lett 4(14): 2329-2332.
- Gerlach M, Jutzi P, Stasch JP, Przuntek H (1982) Synthesis and pharmalogical properties of 4,4-diphenyl-4-sila-piperidines. Z Für Naturforschung B 37(5): 657-662.
- Peters M, Hallensleben ML, Van Hooren M (1999) Synthesis and electrochemical investigations of crown-ether-functionalised bipyrroles. J Mater Chem 9: 1465-1469.
- Zhou HH, Shang XM, Luo Z, Xia YX, Luo J, et al. (2009) A new member of the calix [4] crown family: Facile synthesis and characterization of a calix [4] crown-9 cone conformer. Chin Chem Lett 20(2): 143-146.
- Xia YX, Zhou HH, Yin Y, Qiu N, Luo J, et al. (2010) Intramolecular cyclization strategy: Synthesis of 1,3- and 1,2-calix [4] crown-7 and calix [4] crown-9 cone conformers. J Incl Phenom Macrocycl Chem 68: 423-429.
- Itahara T (1980) Dimerization of pyrroles by palladium acetate. new synthesis of 2,2′-bipyrroles. J Chem Soc Chem Commun 2: 49b-50.
- Kauffmann T, Lexy H (1981) Übergangsmetallaktivierte organische Verbindungen, X: Synthese und Eigenschaften von all-α-Poly(N-methylpyrrolen). Chem Ber 114: 3674-3683.
- Geier GR, Grindrod SC (2004) Meso-substituted [34] octaphyrin(1.1.1.0.1.1.1.0) and corrole formation in reactions of a dipyrromethanedicarbinol with 2,2‘-bipyrrole. J Org Chem 69(19): 6404-6412.
- Henry R, Neal C (1962) 2,2″-bipyrrole. J Am Chem Soc 84(11): 2178-2181.
- Xiao Z, Zhou B, Wang J, Zuo C, He D, et al. (2019) PEO-based electrolytes blended with star polymers with precisely imprinted polymeric pseudo-crown ether cavities for alkali metal ion batteries. J Membr Sci 576: 182-189.
- Kauffmann T, Legler J, Ludorff E, Fischer H (1972) Synthese und eigenschaften von azol-pyridin-kombinationen; Problem der hydrolytisch spaltbaren hetaren-kombinationen. Angew Chem 84: 828-829.
- Clauson Kaas N, Tyle Z, Rottenberg M, Stenhagen E, Östling S (1952) Preparation of cis- and trans 2,5-dimethoxy-2-(acetamidomethyl)-2,5-dihydrofuran, of cis- and trans 2,5-dimethoxy-2-(acetamidomethyl)-tetrahydrofuran and of 1-phenyl-2-(acetamidomethyl)-pyrrole. Acta Chem Scand 6: 667-670.
- Azizi N, Khajeh Amiri A, Ghafuri H, Bolourtchian M (2011) Toward a practical and waste-free synthesis of thioureas in water. Mol Divers 15(1): 157-161.
- Heinze J, Frontana Uribe BA, Ludwigs S (2010) Electrochemistry of conducting polymers--persistent models and new concepts. Chem Rev 110(8): 4724-4771.
- Randriamahazaka H, Sini G, Tran Van F (2007) Electrodeposition mechanisms and electrochemical behavior of poly(3,4-ethylenedithiathiophene). J Phys Chem C 111(12): 4553-4560.
- Asavapiriyanont S, Chandler GK, Gunawardena GA, Pletcher D (1984) The electrodeposition of polypyrrole films from aqueous solutions. J Electroanal Chem Interfacial Electrochem 177(1-2): 229-244.
- Skompska M, Chmielewski MJ, Tarajko A (2007) Poly(1,8-diaminocarbazole) – a novel conducting polymer for sensor applications. Electrochem Commun 9(4): 540-544.
- Skompska M, Vorotyntsev MA, Refczynska M, Goux J, Lesniewska E, et al. (2006) Electrosynthesis and properties of poly(3,4-Ethylenedioxythiophene) films functionalized with titanocene dichloride complex. Electrochimica Acta 51(11): 2108-2119.
- Diaz AF, Martinez A, Kanazawa KK, Salmón M (1981) Electrochemistry of some substituted pyrroles. J Electroanal Chem Interfacial Electrochem 130: 181-187.
- Diaz AF, Castillo J, Kanazawa KK, Logan JA, Salmon M, et al. (1982) Conducting poly-n-alkylpyrrole polymer films. J Electroanal Chem Interfacial Electrochem 133(2): 233-239.
- Rohde N, Eh M, Geißler U, Hallensleben ML, Voigt B, et al. (1995) Oligo(1-methylpyrrole) s-a model system in the conducting polymer series. Adv Mater 7(4): 401-404.
- Qiu YJ, Reynolds JR (1992) Electrochemically initiated chain polymerization of pyrrole in aqueous media. J Polym Sci Part Polym Chem 30(7): 1315-1325.
- Fakhry A, Cachet H, Debiemme-Chouvy C (2013) Electrochemical characterisations of ultra-thin overoxidized polypyrrole films obtained by one-step electrosynthesis. J Electrochem Soc 160(10): D465.
© 2024 Wolfdietrich Meyer. This is an open access article distributed under the terms of the Creative Commons Attribution License , which permits unrestricted use, distribution, and build upon your work non-commercially.
Editor In Chief







.jpg)

























Signup for Newsletter

Quick Links
 Editorial Board Registrations
Editorial Board Registrations Submit your Article
Submit your Article Refer a Friend
Refer a Friend Advertise With Us
Advertise With UsOur Recent Edition

.jpg)



Top Editors




.jpg)

















.bmp)
.jpg)
.png)
.jpg)













.jpg)






.png)

.png)




.png)




Financial Support

Sponsors

Latest e-Books

Latest Video

 a Creative Commons Attribution 4.0 International License. Based on a work at www.crimsonpublishers.com.
Best viewed in
a Creative Commons Attribution 4.0 International License. Based on a work at www.crimsonpublishers.com.
Best viewed in