
- About Us
- Information
-

The Author ensures that the research has been conducted responsibly and ethically with adherence to all relevant regulations. read more..
- For Authors
- For Reviewer
- Manuscript Guidelines
- Membership
- Publication Ethics
-
- Journals
- Reprints
- e-Books
- Videos
- Policies
- Contact Us
 COVID-19
COVID-19
- Submissions



Full Text
Progress in Petrochemical Science
Machine Learning Assisted Material Design Accelerating Progress in Petrochemical Science: Designing Materials for CO2 Photo Capture
Junaid Shahzad*
Department of Chemical Engineering, School of Chemical and Materials Engineering, National University of Sciences and Technology, Pakistan
*Corresponding author: Junaid Shahzad, Department of Chemical Engineering, School of Chemical and Materials Engineering, National University of Sciences and Technology, Islamabad, Pakistan
Submission: November 21, 2022;Published: February 13, 2023

ISSN 2637-8035Volume5 Issue1
Abstract
Carbon Dioxide (CO2) reduction to value-added chemicals, including alcohols and light olefins via heterogeneous catalysis utilizing novel photocatalysts, can reduce the adverse effects of excessive CO2 emissions and our dependence on fossil fuels. In2O3, Cu, Fe and Zn based catalysts coupled with zeolites can increase the selectivity of desired products. Computational techniques such as density functional theory and machine learning assisted materials design will enable versatile and robust photo capture of CO2. Utilizing machine learning models unlocks the potential to discover catalysts, predict catalyst performance, aid in the prediction of catalyst stability, and target performance enhancements of catalysts to further improve the selectivity of desired products.
Keywords: Carbon dioxide; Alcohols; Olefins; Heterogeneous; Photocatalysts; Fossil fuels
Introduction
The surge in the concentration of CO2 in the atmosphere has sparked the interest of research groups in finding sustainable solutions. The conversion of CO2 to renewable fuels, utilizing it as an alternative feedstock for synthesizing value-added materials, is of significance, as this conversion of CO2 produced from the exhaust streams of energy production processes addresses both energy security and climate change [1]. The concept of utilizing solar energy to generate fuels originated from research focused on hydrogen generation from water. The analogy is based solely on the products formed during natural photosynthesis, hence named “artificial photosynthesis,” and the materials which enable or catalyze such processes are called photocatalysts [2]. Photocatalysts have been utilized for the reduction of CO2 sustainably. To scale the process, multiple cells can also be connected in series. Careful investigation of CO2 activation on surfaces may provide insight for designing materials that will power these solar fuel technologies in the future. The term “solar fuels” has already been used to describe former processes. However, recently, the reduction of CO2 has also come to be known as “reverse combustion” [3].
CO2 Reduction Mechanism
Research has been focused on CO2 reduction, explaining its mechanism in systems utilizing metallic electrodes. While the use of semiconductors for CO2 reduction has been reported as early as the late 1970s, metallic electrodes for CO2 reduction were also discovered during the same time [4]. Semiconductors can utilize solar energy to complement or even substitute electrical energy inputs [5]. Semiconductor electrodes vary from metal electrodes as the applied voltage does not affect its electrochemistry directly. Metallic electrodes vary from semiconducting electrodes in the band structure. In metals, the overlapping bands produce a continuum of energy levels for electrons, allowing them to display significant electrical mobility due to the absence of a band gap. In Semiconductors, the band gaps are separated by a quantum mechanically forbidden energy zone, the large energy spacings between the two bands, which results in the valence band being virtually full while the conduction band is empty owing to the higher energy necessary for the transition between the valence band and the conduction band. Electrical mobility is generated by stimulating the electrons between the two bands with photons of energy greater than or equal to the energy required for the transition between the valence band and the conduction band. Photogenerated electrons from the valence band are stimulated to the conduction band, leaving behind positively charged hole-like states in the valence band. Electrons and holes produced by photosynthesis may be exploited in electrochemical processes [6]. The reactions progress as methanol (MeOH) is synthesized from CO2 and H2 over a photocatalyst which may be In, Cu, Fe, or Zn. The dehydration of MeOH is accompanied by zeolites to Dimethyl-Ether (DME), gasoline, and diesel.
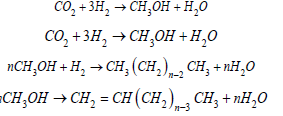
Photocatalysts for CO2 Reduction
Apart from the materials, the dimensionality, topology, and exposed crystal surfaces of photoelectrodes all have an impact on their performance. Transforming materials into nanostructures is a well-established approach to increasing the surface area to volume ratio, resulting in a substantial increase in the density of surface-active sites. Second, arrays of nanostructures may enhance photo absorptivity by minimizing incoming reflectivity while concurrently boosting dispersion and secondary absorption among nanostructures. Thirdly, lowering the size of the crystal decreases the distance between minority carrier photo generation and the charge transfer interface, hence minimizing the likelihood of electron-hole recombination. The proportions and design of the surfaces, grain boundaries, and defects of a material change depending on the materials or manufacturing technique utilized to create that material [7]. In2O3-based catalysts have received considerable attention due to their exceptional results and simple active site designs. Furthermore, these catalyst systems generally demonstrate excellent selectivity and high stability [8]. However, their single pass CO2 conversions are considerably lower than those of supported metal catalysts (such as Cu- and Fe-based catalysts) and are therefore typically below 20 %, even with noble metal modifiers and at elevated reaction temperatures (>280 °C) [9]. In industrial applications for CO2 hydrogenation, a rather large recycling ratio of the unconverted gas is needed, which may compromise the process’s energy efficiency and economic value. To raise the intrinsic activity of these oxide catalysts, it is necessary to increase the number of oxygen vacancies and Hydrogen splitting capacity. When the CO2 source is flue gas from coal or biomass combustion, the CO2 conversion process becomes more challenging due to CO, O4, SOx, and NOx presence. The expense of treating flue gas, especially extensive desulfurization, will lower the economic viability of the CO2 conversion process. ZnO-ZrO2 catalysts are more suited due to their greater sulphur tolerance [10].
Design of Heterogeneous Photo Catalysts for CO2 Reduction
Across several disciplines, including drug discovery, molecular biology, and material and process design, the estimation of molecular characteristics is a crucial task. Scientists initially accomplished the identification and characterization of novel compounds manually and intuitively. Three paradigm shifts have occurred in materials science, including experimental research, mathematical theory, and simulation [11]. The first paradigm in which the study of material science depended only on intuitive observation experience and lacked any scientific foundation for quantification. In the second paradigm, physical models described by mathematical equations began to emerge, providing some theoretical background for materials research, such as thermodynamic principles [12]. The introduction of computers ushered in the third scientific paradigm, which enabled the modeling of complicated practical issues based on the second paradigm’s theory. During this time, two critical methodologies, density functional theory, and molecular dynamics were developed for materials research using big data computing. Using computational means, the Quantitative Structure-Property Relationship (QSPR) modeling technique has established a valuable and efficient framework for screening and exploring the chemical search space. QSPRs describe molecules using multiple structural, chemical, physical, and biological characteristics, known as molecular descriptors, which are subsequently mapped to an essential attribute via a linear or nonlinear model [13,14], (Figure 1). Nevertheless, selecting valuable descriptors is a challenging task that often requires domain expertise and expert intuition, with a poor choice resulting in poor prediction performance. However, the study procedures of conventional materials science, which depend primarily on trial-and-error techniques, usually take 15-25 years or even more from research to application, and countless inaccurate conclusions have not been implemented [15]. Due to the vast quantities of data and high dimensions in materials research, more and more material characterization methods are being developed. Even though the structures and performance of materials can be calculated and predicted at different scales using material simulation methods such as first principles, molecular dynamics, phase-field theory, and finite element analysis, the models are typically material system-specific. It is still incapable of meeting the criteria of several descriptions for various qualities, severely limiting the scope of future material research [16].
Figure 1A suggested framework for training, development, and evaluation of a machine learning model capable of material design and evaluation.

Machine Learning Assisted Material Design
Recent developments in computing technology and software have enabled the development of deep learning, including novel Machine Learning (ML) algorithms that have found tremendous success in various domains, including pattern recognition and natural language processing. Deep learning may automatically discover fundamental representations from given data instead of depending on expert judgment for feature engineering. This has also resulted in a revival and subsequent improvements in machine learning-based property prediction and molecule production. Random Forest (RF) is based on constructing a training set and constructing a predictor tree that maps out observations of a given variable to produce accurate and reliable predictions of the target. RF is more superficial to read than ANN because of its simplicity and clarity. It classifies a data point from the top of the tree through different branches, down through the various nodes, and to the terminal leaf. In addition, they provide additional information on the relative significance of descriptors in terms of how they impact the target. In addition, all ML approaches have their benefits and limitations, and the selection of a particular method is contingent upon the size and characteristics of the database. For small data sets, linear regression, and classification algorithms, such as the Logistic and Naive Bayes methods, may handle such jobs. On the other hand, nonlinear approaches, such as ANN and K Nearest- Neighbor (KNN) algorithms, are suitable for large data sets. Due to its easy-to-learn nature, the ANN technique often yields superior initial model performance with larger data sets.
Conventional techniques for material design are based on experimentation and are led by intuition, experience, and conceptual insights. Given their successful use in discovering several critical materials, these techniques face complex challenges due to the enormous need for more efficient technology and the enormous potential for novel material development. We can systematically identify new materials by developing algorithmic tools for designing, synthesizing, and discovering materials that may have significant technological and social impacts. A comprehensive evaluation of the multistage design process, including exploration of vast material spaces, their characteristics, and their design and engineering, is necessary for these rational designs. The complexity of using rigorous experimental methods to investigate these possibilities seems unachievable. Machine Learning (ML)- assisted materials design is developing as a prospective instrument to create advances in several fields of materials design because of breakthroughs in computational methods, artificial intelligence (particularly Machining Learning, ML), and the creation of materials databases. The quantitative structural property/activity connection for material property prediction may now be developed more accurately and effectively using ML-assisted techniques. The ability of Machine learning algorithms to comprehend the syntax of chemically relevant representations of molecules allows for the creation of novel materials. The majority of scientific discoveries throughout history have not been made only based on logic but rather on the base of intuition. Researchers might be seen in this context as intelligent decision-making “black boxes” with strong intuitions. Machine learning algorithms appeal to more researchers because, after discovery or prediction, researchers can always go back and look at the decision-making processes to better understand. Consequently, we can now target tailoring material properties and the future of material design by leveraging the possibilities that machine learning presents for doing better research. In order to address the enormous demand for novel materials with specialized features, it may be predicted that ML-assisted materials design will become more critical in the near future.
References
- Ghuman KK, Wood TE, Hoch LB, Mims CA, Ozin GA, et al. (2015) Illuminating CO2 reduction on frustrated lewis pair surfaces: Investigating the role of surface hydroxides and oxygen vacancies on nanocrystalline In2O3-x(OH)y. Physical Chemistry Chemical Physics 17(22): 14623-14635.
- White JL, Baruch MF, Iii JEP, Hu Y, Fortmeyer IC, et al. (2015) Light-driven heterogeneous reduction of carbon dioxide: Photocatalysts and photoelectrodes. Chemical Reviews 115(23): 12888-12935.
- Kumar B, Llorente M, Froehlich J, Dang T, Sathrum A, et al. (2012) Photochemical and photoelectrochemical reduction of CO2. Annu Rev Phys Chem 63: 541-569.
- Hori YI (2008) Electrochemical CO2 reduction on metal electrodes. Modern Aspects of Electrochemistry 42: 89-189.
- Halmann M (1978) Photoelectrochemical reduction of aqueous carbon dioxide on p-type gallium phosphide in liquid junction solar cells. Nature 275: 115-116.
- Bard AJ, Faulkner LR (2001) Electrochemical methods: Fundamentals and applications p. 864.
- Yu M, Long YZ, Sun B, Fan Z (2012) Recent advances in solar cells based on one-dimensional nanostructure arrays. Nanoscale 4(9): 2783-2796.
- Gao P, Zhang L, Li S, Zhou Z, Sun Y (2020) Novel heterogeneous catalysts for CO2 hydrogenation to liquid fuels. ACS Central Science 6(10): 1657-1670.
- Macdowell N, Florin N, Buchard A, Hallett J, Galindo A, et al. (2010) An overview of CO2 capture technologies. Energy & Environmental Science 3(11): 1645-1669.
- Goeppert A, Czaun M, Jones JP, Prakash GKS, Olah GA (2014) Recycling of carbon dioxide to methanol and derived products-closing the loop. Chemical Society Reviews 43(23): 7995-8048.
- BO Gregory (2000) Designing a material world. Science 288: 993-998.
- Luo Q, Guo Y, Liu B, Feng Y, Zhang J, et al. (2020) Thermodynamics and kinetics of phase transformation in rare earth-magnesium alloys: A critical review. Journal of Materials Science & Technology 44: 171-190.
- Boyd PG, Lee Y, Smit B (2017) Computational development of the nanoporous materials genome. Nature Reviews Materials 2(8): 1-15.
- Wu X, Kang F, Duan W, Li J (2019) Density functional theory calculations: A powerful tool to simulate and design high-performance energy storage and conversion materials. Progress in Natural Science: Materials International 29(3): 247-255.
- Hou J, Chen M, Zhou Y, Bian L, Dong F, et al. (2020) Regulating the effect of element doping on the CO2 capture performance of kaolinite: A density functional theory study. Applied Surface Science 512: 145642.
- Kunwar A, An L, Liu J, Shang S, Råback P, et al. (2020) A data-driven framework to predict the morphology of interfacial Cu6Sn5 IMC in SAC/Cu system during laser soldering. Journal of Materials Science & Technology 50: 115-127.
© 2023 Junaid Shahzad. This is an open access article distributed under the terms of the Creative Commons Attribution License , which permits unrestricted use, distribution, and build upon your work non-commercially.
Editor In Chief







.jpg)

























Signup for Newsletter

Quick Links
 Editorial Board Registrations
Editorial Board Registrations Submit your Article
Submit your Article Refer a Friend
Refer a Friend Advertise With Us
Advertise With UsOur Recent Edition

.jpg)



Top Editors




.jpg)

















.bmp)
.jpg)
.png)
.jpg)













.jpg)






.png)

.png)




.png)




Financial Support

Sponsors

Latest e-Books

Latest Video

 a Creative Commons Attribution 4.0 International License. Based on a work at www.crimsonpublishers.com.
Best viewed in
a Creative Commons Attribution 4.0 International License. Based on a work at www.crimsonpublishers.com.
Best viewed in