
- About Us
- Information
-

The Author ensures that the research has been conducted responsibly and ethically with adherence to all relevant regulations. read more..
- For Authors
- For Reviewer
- Manuscript Guidelines
- Membership
- Publication Ethics
-
- Journals
- Reprints
- e-Books
- Videos
- Policies
- Contact Us
 COVID-19
COVID-19
- Submissions



Full Text
Progress in Petrochemical Science
The Direct Dimethyl Ether (DME) Synthesis Process from Carbon-Based Feed Stocks: Current -Status and Future Prospects II. Kinetic Studies and Catalyst Deactivation
Makarand R Gogate*
Jawaharlal Nehru College of Engineering, India
*Corresponding author: Makarand R Gogate, Jawaharlal Nehru College of Engineering, 259 Samarthnagar, Opp SBI Branch, Aurangabad, India
Submission: May 11, 2018;Published: August 16, 2018

ISSN 2637-8035Volume2 Issue4
Abstract
In Part I of this series, it was seen that the favorable thermodynamic and kinetic coupling in the LPDMEtm process--of methanol dehydration reaction (very rapid and at/near thermodynamic equilibrium) with the methanol synthesis reaction (slower kinetics and highly thermodynamic) --leads the beneficial “chemical synergy”. This synergy helps to overcome the limitation on thermodynamic equilibrium conversion and increases the per-pass syngas conversion and reactor productivity. This increase in the per-pass syngas conversion can be as high as 50-100% and depends primarily on the feed H2: CO ratio.
In this part II of Series, we briefly discern the intrinsic kinetics of the LPMeOHtm and LPDMEtm systems, and also shed light of the catalyst deactivation phenomena in these processes. Among the many reports on intrinsic kinetics of the one-step LPMeOHtm and LPDMEtm processes, two illustrative kinetic studies, from the groups of University of Akron and Air Products and Chemicals, Inc. are highlighted and discussed further. These are mainly based on the independent, component kinetic models of methanol synthesis (Vanden Bussche and Froment) and methanol dehydration (Bercic & Levec). From an overarching analysis of the deactivation of supported copper catalysts for methanol synthesis and other reactions (methanol decomposition and methanol steam reforming), we propose that thermal sintering, i.e., increase in Cu particle site and loss of metal surface area, is the only cause of catalyst deactivation in methanol synthesis reactions over Cu/ZnO/Al2O3 industrial-type methanol catalysts. In closing, we outline the reactor design/scale-up and plant operational experience of the 3 commercial technologies, as currently practiced by JFE holdings, BP-AMOCO, and Halder-Topsoe.
keywords:Natural gas; Steam reforming; Coal; Syngas; Methanol; DME; Bi-functional catalysts; Cu/ZnO/Al2O3; γ-Al2O3; Slurry reactors; Bubble column reactors; Chemical synergy; Methyl equivalent productivity (MEP); Intrinsic kinetics; Phase equilibrium; Chemical reaction equilibrium; Catalyst deactivation
Introduction
In Part I of this series; we illustrated the process feasibility of the LPMeOHtm and LPDMEtm processes and shed an illuminating light on the chemical synergy and role of the water gas shift reaction in the overall process chemistry. These 2 processes have been illustrative examples of how a mature technology on a commercial scale (ICI low temperature methanol synthesis process) can be successfully adapted to a liquid phase operation. It is possible that this adaptability and flexibility of a cost-effective liquid phase alternative can be extended to other parallel reactions; involving multiple components; especially if the overall reaction is highly exothermic and reversible. The LPMeOHtm process is a highly flexible process that is well-suited to syngas feeds of all H2: CO ratios (from H2-rich to CO-rich); uses milder reactor design conditions (of temperature and pressure); and a simpler one from a process engineering stand point. Some of the key advantages of the LPMEOHtm process include better heat transfer characteristics and isothermal operation; use of the CO-rich syngas (from low-cost coal sources); and a very high chemical selectivity to methanol.
The process chemistry of LPDMEtm system is covered in detail in Part I of this Series. The process is based on application of dual catalysis in a single reactor stage; and based on a combination of an equilibrium-limited reaction (methanol synthesis) and an equilibrium unlimited reaction (methanol dehydration). The selective chemical removal of product methanol; from Reaction (1); via its dehydration; via Reaction (3); overcomes the chemical equilibrium barrier on methanol synthesis alone; and improves the per-pass syngas conversion and reactor productivity.
Kinetic Studies on LPMEOHtm and LPDMEtm Processes
In this Part II; we focus on the kinetic studies of the liquid phase methanol; LPMeOHtm and direct one-step DME synthesis from syngas; the LPDMEtm process. The kinetic studies often have important implications for process design; scale-up; and commercial operation. The literature studies on the intrinsic kinetics can be classified into 3 distinct categories:
a) Kinetics of vapor phase synthesis of methanol from CO/ CO2 and H2 (ICI low pressure methanol synthesis; over Cu/ZnO/ Al2O3 catalyst)
b) Kinetics of liquid phase methanol from CO/CO2 and H2over Cu/ZnO/Al2O3 (LPMeOHtm process; by Chem Systems; Inc.)
c) Intrinsic kinetics of the direct; one-step DME process (LPDMEtm process; which was introduced by UA researchers’; in 1989)
Here we will only consider the state-of-art literature on kinetic studies on the liquid phase methanol and liquid phase DME (LPDMEtm) processes and provide a critical assessment of proposed kinetic rate expressions; and discern the benefits and drawbacks of our current understanding of the literature on intrinsic and in-situ kinetics. It is interesting to note that the vapor phase mechanisms and proposals for kinetic rate expressions are more difficult to discern than the liquid phase systems because of the differences in reactor designs/operational conditions in differential or pseudo-differential modes (which can confound data analysis and comparison to integral modes of operation); a problem not encountered in the liquid phase mode of operation.
For development of intrinsic kinetic models of LPMeOHtm and LPDMEtm systems; it is first necessary to develop a detailed thermodynamic framework of this multi-component; multiphase system; where at least 8 independent components participate in 3 parallel reactions (methanol synthesis from CO/CO2; water-gas shift; and methanol dehydration); over solid catalysts; well-dispersed in Witco-40 inert mineral oil.We illustrate briefly the computational procedure to develop the multi-component phase and chemical reaction equilibria models for LPMeOHtm and LPDMEtm processes. We refer to the interested reader a few seminal reports from the researchers at University of Akron [1-3]. Regardless of the kinetic model and the type of formalism chosen to describe it (L-H; power law; etc.); it is first necessary to compute the concentrations (and fugacities) of all species; i.e.; reactants and products; in the liquid phase. To this end; extensive experimental studies on the binary and multi-component phase equilibria of syngas components at a range of pressures and temperatures; to cover the range of practical interest; were carried out by UA researchers’. The data from the binary experiments was incorporated into a multi-component phase equilibria model-the Margule’s equations were used to model the binary vapor-liquid equilibria and to generate the Margule’s parameters; using Lewis-Randall standard state for vapors/liquids (CH3OH; H2O; DME; Witco-40); and the Soave-Redlich-Kwong equation of state (EOS) was used to model the multi-component phase equilibria; with Henry’s law constants as standard states for permanent gases (H2; CO; CO2; CH4). A complete computer program package (in FORTRAN-90) was developed by the UA research group; that calculates concentrations of all reactive species in the liquid phase; based on the simultaneous multi-component phase and multi-reaction chemical equilibria [1-3]. As input data; only the vapor phase compositions of all species (from experimental data); pressure; temperature; and volume of oil are required. The system of non-linear algebraic equations with 8 unknowns (coupled) is solved using the Newton-Raphson iteration method; with Gauss- Jordan reduction algorithm for computation of inverse Jacobians; for rapid convergence. The detailed calculations of thermodynamic equilibrium constants for the 3 principal reactions; Kf;A; Kf;B; and Kf;C; as functions of T; are readily available in the literature [4-9].
The first results on the kinetic studies on the LPMeOHtm process were presented by the APCI group at the International Symposium of Chemical Reaction Engineering (popularly given as I.S.C.R.E.); in Edinburgh; Scotland; in 1984 [10] and subsequently presented in a more refined form at 2 other Symposia/Conferences [11,12]. Later; researchers’ from APCI presented their kinetic models in a Langmuir-Hinshelwood form. The APCI work was also presented in another form-a modified L.H. form-also including an activity factor; A; to account for catalyst deactivation; i.e.; to allow for the gradual loss in activity as a function of time on stream [13-17]. An overall analysis of APCI kinetic studies indicates that the apparent activation energy is in the range of 75,000-90,000J/mol; and the pre-exponential factors are of the order of kp;o=2.43x104mol CH3OH/s.kg cat. MPa; and for the kinetic model based on fugacity terms; kf;o=3.68x106mol CH3OH/h.kg cat.atm. As an anecdotal note; Sherwin & Blum of Chem Systems; Inc; proposed the very first kinetic rate expression of LPMeOHtm process; as a power law form; but incorrectly used the CO-COeq term as a variable for the functional form (and not CH2-CH2;eq). It is interesting to note that almost all kinetic rate expressions on liquid phase methanol seem to overwhelmingly agree that CO exhibits a very weak dependence on methanol rates-the reaction order with respect to CO is often found to be close to zero. At the same time; three independent research groups; UA (Akron; Ohio); University of Aachen/Groningen (Germany/Netherlands); and Technical University of Lodz (Poland) also proposed the kinetic rate expressions for the LPMeOHtm process. For example; the Lodz research group [18] proposed a kinetic rate expression written as the difference between a forward kinetic term and a backward kinetic term (for the reverse reaction):
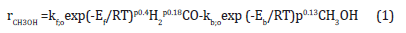
The values of Ef and Eb; according to the kinetic model; were given to be 56000J/mol and 85000J/mol.The forward and backward kinetic terms were given as 5.5x103 and 6x103mol CH3OH/sec. kg cat. MPa-2 (The negative exponent on the MPa unit; of -2; indicates the value of Δn; or; change in number of moles). The low estimates of reaction orders; 0.4 for H2 and 0.18 for CO; indicate that they are likely not true reaction orders; but apparent ones; likely influenced by external mass transfer (due to high slurry ratios); or even pore diffusional issues. The particle size; of 60μm; is rather high. The work of Von Wedel et al. [18] was extended by Ledakowicz et al. [19]; who proposed a kind of hybrid kinetic model-a power law combined with elements of L-H formalism. In contrast with the works above and the extensive kinetic analysis of Graaf et al. [4,5] that follows; based on a detailed micro-kinetic model of methanol synthesis reaction; a relatively simple kinetic analysis was presented by the UA research group [1]; for liquid phase methanol process; over commercial Cu/Zn/Al catalyst of United Catalysts; Inc. The rate of methanol synthesis was found to be independent of partial pressures of CO; CO2 and H2O. Consequently; the methanol rate was modeled only as a function of the concentration driving force (approach to equilibrium) of H2; as follows:

A statistical regression analysis of the rate data as a function of the H2 driving force showed that the reaction order; n; ranged from 0.80 to 1.15; and could be approximated well by the value of 1; over the entire range. This model; although simple; is in good agreement with all literature results; which suggest that methanol synthesis rates exhibit a very weak dependence on concentrations of CO in the liquid phase. An extended kinetic model for the liquid phase methanol process was developed by [4,5]. Their experimental component was carried out in a 300cm3 stirred autoclave; with a very low catalyst loading; to ensure absence of external gas-toliquid mass transfer limitation. For the three principal reactions; CO hydrogenation; water-gas shift (CO2+H2); and CO2 hydrogenation; a detailed micro-kinetic model based on series of elementary surface reactions was proposed. Based on a series of 4 elementary reactions for (A); 2 reactions for (B); and 6 reactions for (C); a total of 48 (4x2x6) kinetic models could be constructed and were considered; for further analysis. Based on experimental data; statistical analysis (χ2-test; with 95% C.I.s; and analysis of error residuals); and physico-chemical constraints; a model termed as A3-B2-C3 was proposed to be the best fit for the kinetics of liquid phase methanol process. The typical L-Hforms of kinetic rate expressions are given in [4,5]. For the APCI portfolio; we begin by considering “seminal” works from the APCI research; both from 1999 [20,21]. The work of Peng et al. [21] is also the most specific in regards the intrinsic kinetics of direct; one-step DME synthesis in slurry mode of operation. For all kinetic studies; the reactor was considered to behave as a CST Rand was free of external mass transfer (external to liquid) limitations and pore diffusional limitations. All three expressions are power law form multiplied by an approach-toequilibrium term as shown below:
a. Methanol synthesis reaction:

b. Forward water-gas shift:
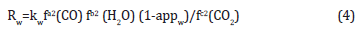
c. Methanol dehydration reaction:

The models have excellent predictive capability and the methyl equivalent productivities (MEPs) as predicted from the kinetic models are in good agreement with experimental results from the data gathered at conditions as above (Figure 1) [21]. In fact; the models have excellent predictive power across the range of syngas types; or H2: CO inlet ratios; for the 3 typical syngas feeds; viz.; Texaco (Koppers-Totzek); the most typical composition of COrich syngas; Shell (similar to Texaco); and Dow (close to H2-rich type).The second report from APCI research by Peng et al. [21] is on intrinsic kinetics and modeling of the direct; one-step DME process; in the vapor phase. Ng et al. investigated the kinetics of the one-step DME synthesis process over a commercial CuO/ZnO/ Al2O3 methanol catalyst and a γ-alumina catalyst at 250 ᵒC and 5MPa; using a gradient-less; internal-recycle-type reactor (Bertytype). A kinetic model for the combined synthesis methanol and DME (co-production of methanol and DME); based on a methanol synthesis kinetic model proposed by Vanden Bussche and Froment (1996) and a methanol dehydration kinetic model by Bercic and Levec (1992) was tested using experimental results obtained in the Berty-type reactor from a wide range of syngas compositions and CO2/CO ratios.
The kinetic parameters of the work of peng et al. [21] are summarized in Figure 1. The proposed kinetic models of peng et al. [21] and the kinetic parameters that are derived from the best fits to the experimental data are given in (Figure 1-3); Table 1; respectively. The illustration of chemical synergy in the LPDMEtm process system (methanol+DME) vs. LPMeOHtm alone is given in Figure 2. The chemical synergy is a very strong function of the H2: CO ratio and the fraction of CO2in the feed syngas. Finally; it is worth noting that most kinetic models proposed in the literature are based on direct CO2 hydrogenation via formate as the intermediate [22,23]. The 3 customarily proposed reaction mechanisms for direct CO2 hydrogenation comprise of formate; formyl (via RWGS reaction); and carboxyl. The three mechanisms are illustrated in Figure 2.
Table 1:Kinetic rate parameters for the rate models given in Figure 1.

figure 1:Kinetic rate models for methanol synthesis reaction via CO2 hydrogenation, water gas shift, and methanol dehydration reaction.

figure 2:The extent of chemical synergy in LPDMEtm vs. LPMeOHtm alone in terms of the effect of GHSV (h-1) on the methanol equivalent yield (MEY), at 250 ᵒC and 50 bar and with a gas composition of 18% COx, 72% H2, and 10% He.

figure 3:Surface reaction mechanisms and reactive intermediates for direct CO2 hydrogenation to methanol over Cu catalysts.

Catalyst Deactivation in LPMeOHtm and LPDMEtm Process
An important distinction needs to be made at the very outset here; between vapor and liquid phase modes of operation. For methanol synthesis; this distinction is more pertinent; as the one of the primary reasons for the liquid phase methanol synthesis process (LPMeOHtm) was introduced by Chem Systems (now Nexant) in 1975 was to control the very highly exothermal nature of the commercial ICI vapor phase methanol process; and thus reduce the thermal sintering phenomena. In this section; we will make only a brief but important comment on the long-term activity patterns (vs. time) and catalyst stability and will focus only the catalyst deactivation phenomena for LPMeOHtm and LPDMEtm processes and elucidate how the presence of the liquid phase is able to moderate the exothermal nature of methanol synthesis reactions; and; in fact; extend on-stream catalyst lifetimes. For sake of completeness; we refer the interested reader to the following papers on the catalyst deactivation of bi-functional catalysts for direct; one-step DME vapor phase process and the catalytic dehydration of methanol to DME [24-33].
An interesting question that has always been confounding to researchers is the nature of the catalyst deactivation--of individual micron-sized Cu/Zn/Al methanol component and the most common γ-Al2O3 or H-ZSM-5 DME component-in the LPDMEtm process; when used in the form of a physical mixture (also called an admixture) and dispersed in an inert mineral oil (like Witco-40; Witco-70; or Freezene-100); at typical operating conditions of 250 ᵒC and 70atm; as a function of on-stream time. It is intuitively obvious; however; that the thermal sintering of the copper component-- most often proposed to be cause of catalyst deactivation in the vapor phase and validated by many studies; including most notably those of [34-36], Kung [24] will be greatly alleviated in the liquid phase operation; as the presence of the liquid phase (with its high heat capacity)serves to moderate the strongly exothermal nature of methanol synthesis reactions; and the presence of “hot spots”; in particular. As the finely dispersed copper component is completely wetted/filled (external and internal surface of pores) by the liquid phase; the reaction is essentially “isothermal”; with no local heat gradients or “hot spots”. The mechanism of thermal sintering that leads to an increase in size of copper crystallites-with the increase in surface area and a concomitant decrease in methanol rates and TOFs-is thus vastly “suppressed” in the liquid phase.
The presence of in-situ produced CH3OH and H2O within the pores of Cu/Zn/Al and γ-Al2O3 is often intuitively considered to be the cause of hydrothermal sintering (i.e.; contribute to the “thermal” sintering effects) and even leaching of the copper and zinc components. Obviously; it is more difficult to remove H2O from the surface of Cu-based catalyst in a slurry reactor configuration; as the surface is within the pore structure of the Cu/ZnO catalyst; completely wetted by the inert liquid phase. This liquid phase poses an additional barrier for a facile back-diffusion of products; CH3 OH and H2O; back into bulk liquid and into vapor phase. However; we do not agree with the hypothesis of hydrothermal leaching or loss of crystallinity as primary causes of deactivation of Cu and Zn crystalline phases.
Our seminal works at University of Akron in Prof. Lee’s group with A. Sawant and B. Lee as doctoral candidates [37-44] and other works conclusively prove that the hydrothermal sintering that leads to a crystal size growth of Cu and the concomitant loss of Cu surface area are the only plausible and possible causes of catalyst deactivation in this phenomena; and the physicochemical interaction between the 2 components is not just benign but can be synergistic. In fact; it is quite apropos to state here that the insitu processes developed by the UA researchers’ for regeneration of aged copper catalysts during LPMeOHtm processes are based on the principle of “crystallite re-dispersion”; i.e; size-reduction accompanied re-dispersion of crystallite sizes. These processes have been demonstrated under bench and pilot scale conditions and even commercial scale operation; at the commercial-scale LPMeOHtm and LPDMEtm processes at Eastman Chemical Co’s plant site in Kingsport; TN; and can restore completely the original particle size distribution of Cu crystallites and the initial activity. The applicability of catalyst regeneration process for aged (deactivated) catalysts based on the principle of crystallite redispersion underscores our assertion of the primary governing mechanism for deactivation and aging; i.e.; thermal sintering and crystal growth that leads to a loss in surface area of active copper.
It is interesting to note that the fact that thermal sintering is the primary mechanism of catalyst deactivation in Cu catalysts in the LPMeOHtm and LPDMEtm processes has been asserted and corroborated by other research reports [45,46]. However; other causes including presence of trace carbonyls such as Fe(CO)5 and Ni(CO)4; carbon deposition by reverse boudouard reaction; and hydrothermal leaching by the high water concentrations in the slurry; can also be present to a smaller extent and can be important. The poisoning by carbonyl sulfide-a compound of CO and sulfur-can also is more significant in the liquid phase processes. These causes can be considered to be minor and can be eliminated to a great extent by good plant design/engineering practice.An interesting mechanism of catalyst deactivation in dual catalysis; or LPDMEtm process; was proposed by Peng et al. [21] as detrimental physical interaction of methanol and DME catalyst components. The data as reproduced in Figure 4 is considered to be questionable and thus unreliable.
Commercial Technologies and Future Markets
Di-methyl ether (DME) is being touted for its potential as a very versatile chemical intermediate as well as a clean-burning alternative fuel and a “green” substitute for diesel; as it does not emit any soot or particulate matter (PM) upon combustion in diesel engines [47-50]. DME is a colorless; odorless gas with physical and chemical properties similar to that of “liquefied petroleum gas (LPG)” which is popular in Southeast Asia (India; China; Pakistan; Bangladesh; etc.) and Pacific including Japan; for household cooking and even heating. Like LPG; DME can be sold in standard bottles/ cylinders as a pressurized liquid at about 5atm; and holds a higher volumetric energy density than LPG gas.DME is now exclusively used in aerosol applications in spray propellants. The current world capacity of DME is 1, 50,000 TPA; Japan is one of the key players in the DME technologies and future markets (with annual capacity of 10,000 TPA and projected to increase rapidly). The large-scale implementation of the cleaner-burning DME as a diesel substitute in European markets and elsewhere where diesel is a popular choice for motor vehicles is a matter of crisscrossing debate between automakers and OEMs; local environmental advocacy groups (like Greenpeace); and importantly; the geopolitical and economic considerations. It is worth noting that DME does not really have a “weak” spot on its resume; it is a cleaner burning fuel; with no particulate emissions; no post-combustion soot; has a very high H/C ratio and additional O atom which makes it very clean; and has a high cetane number (equivalent to the octane number for gasoline-boiling fraction; C5-C10); in fact; the volumetric energy value of DME is higher than that of diesel.
figure 4:Normalized activity vs. time on stream for different liquid phase processs. Symbol legend: (Δ), km, methanol synthesis catalyst, via LPMeOHtm only; (x), kd, methanol dehydration only; (●), km, methanol synthesis catalyst, LPDMEtm system; kd, methanol dehydration catalyst, γ-Al2O3O, LPDMEtm system.

The direct; one-step DME process as described in this paper and the various elements that comprise the current research areas on bench-scale and especially at pilot scale (principally at UA and APCI research) has now formally made the transitional debut at the commercial scale; in Hokkaido; Japan.JFE company (previously NKK Nippon Corporation); in collaboration with Taiheiyo Coal Mining Co.; Sumitomo Metal Industry; Ltd.; and CCUJ (Center for Coal Utilization; Japan); have already demonstrated this technology on a 5ton DME/day (5TPD) pilot scale in 2002 [40-42]. Based on this achievement; JFE Co. successfully translated this pilot scale demonstration into a commercial-scale 100 ton DME/day (100 TPD) DME plant at its site in Hokkaido; Japan; starting 2005.
Korea Gas Corporation (KGC); Halder-Topsoe (based in Denmark); Air Products (Allentown; PA; U.S.A.); and BP-AMOCO (Des Plaines; IL; U.S.A.) are also on the commercialization track for the direct; one-step DME process/technology. A Halder-Topsoe patent (WIPO International Patent WO96/23755) outlines their research efforts towards development of the direct; one-step DME process. The status of DME technology development efforts by BP-AMOCO for their licensees in China and other players in Asia are summarized in a review [46]. A totally new and highly innovative “application” of the direct; one-step DME technology; which is inherently simpler and cost-effective (when compared to the conventional process with 2 separate steps) can be thought of in the traditional gas-to-liquids (GTL) market segments. The GTL technologies offer the owners/operators of stranded gas reserves a cheap alternative to ship stranded gas in the form of liquids; which are easier and more economical to transport. However; the technologies are complex; costly; and tightly held by a few major companies; which present significant impediments for new (and independent) large-scale plants; in terms of capital costs and longterm technology transfer decisions. In this regard; a small-scale; direct DME technology modular application; can be ideal for such stranded gas reserves or even for associated gas (typically sent to flares); for local end uses.
Summary
In Part I of this Series; we illustrated the chemical synergy in the LPDMEtm process; and saw how the combination of an equilibriumlimited reaction; methanol synthesis; and an equilibrium-unlimited one; methanol dehydration to form dimethyl ether; can help lift the chemical equilibrium limitation on methanol synthesis alone.
This thermodynamic-kinetic coupling is the basis of the chemical synergy observed in both LPMeOHtm and LPDMEtm processes. In this part II of Series; we have discerned the intrinsic kinetics of the LPMeOHtm and LPDMEtm systems; and also shed light of the catalyst deactivation phenomena in these processes. From an overarching analysis of the deactivation of supported copper catalysts for methanol synthesis and other reactions (methanol decomposition and methanol steam reforming); we have demonstrated that thermal sintering; i.e.; increase in Cu particle site and loss of metal surface area; is the only cause of catalyst deactivation in methanol synthesis reactions over Cu/ZnO/Al2O3 industrial-type methanol catalysts.
The future of DME as an alternative fuel and a chemical intermediate/commodity for targeted end use is very bright. The direct; one-step DME process as described in this paper and the various elements that comprise the current research areas on bench-scale and pilot scale (principally at UA and APCI research in the United States and Halder Topsoe in Denmark) has now formally made the transitional debut at the commercial scale; in Hokkaido; Japan. The JFE Holdings owned commercial-scale DME plant has an on-stream capacity of 300 TPD and has been in operation since 2007.Korea Gas Corporation (KGC); Halder-Topsoe (based in Denmark); Air Products (Allentown; PA; U.S.A.); and BP-AMOCO (Des Plaines; IL; U.S.A.) are also on the commercialization track for the direct; one-step DME process/technology.
References
- Lee S (1990) Methanol Synthesis Technology. CRC Press, Boca Raton, Florida, USA.
- Ko M, Lee S (1987) Multi-component physical equilibrium of liquid phase methanol synthesis process. Energy & Fuels 1(2): 211-216.
- Ko M (1987) Thermodynamic analysis and mass transfer study of liquid phase methanol synthesis process. PhD Dissertation, University of Akron, Akron, Ohio, USA.
- Graaf GH, Winkelman JGM, Stamhuis EJ, Beenackers AACM (1988a) Kinetics of three phase methanol synthesis. Chem Eng Sci 43(8): 2161- 2168.
- Graaf GH, Stamhuis EJ, Beenackers AACM (1988b) Kinetics of low pressure methanol synthesis. Chem Eng Sci 43(12): 3185-3195.
- Beenackers AACM, Graaf GH, Joosten GEH (1987) Recent trends in chemical reaction engineering. In: Kulkarni BD, Mashelkar RA, Sharma MM (Eds.), Wiley Eastern Ltd, New Delhi , India, pp. 45-70.
- Lee S, Berty JM, Greene HL, Desirazu S, Ko M, et al. (1984) Thermodynamics, kinetics, and thermal stability of liquid phase methanol synthesis process. Electric Power Research Institute, California, USA.
- Lee S, Sawant A, Parameswaran V, Sullivan T (1985) Research into thermodynamics, mass transfer, oil and catalyst degradation in liquid phase methanol process. Electric Power Research Institute, California, USA.
- Graaf GH, Sijtsema PJJ, Stamhuis EJ, Joosten GEH (1986) Chemical equilibria in methanol synthesis. Chem Eng Sci 41(11): 2883-2890.
- Brown DM, Gottier GN, Upadhye RS, Bauer JV, Cilen NA, et al. (1984) Modeling of methanol synthesis in the liquid phase. Institution of Chemical Engineers 87: 699-708.
- Klosek, J, Brown DM, Mednick RL (1985) Status of the La Porte LPMe OH process demonstration unit (PDU). Proceedings of Tenth Annual EPRI Contractor’s Conference on Clean Liquid and Solid Fuels, Palo Alto, California, USA, pp. 23-25.
- Weimer RF, Terry DM, Stepanoff P (1987) Laboratory kinetics and mass transfer in the liquid phase methanol process (LPMeOHtm) process. AIChE Fall Annual Meeting, New York City, USA.
- Brown DM, Hsiung TH, Rao P, Greene MI (1985) Catalyst activity and life in LPMeOHtm process. Proceeding of the Tenth Annual EPRI Contractor’s Conference on Clean Liquid and Solid Fuels, Electric Power Research Institute, Palo Alto, California, USA, pp. 23-25.
- Brown DM, Henderson JL, Hsiung TH, Studer DW (1990) LPMeOH: Beyond LaPorte-next steps to commercialization. Proceedings of Fifteenth EPRI Annual Conference on Fuel Science, Electric Power Research Institute, Palo Alto, California, USA, pp. 19-22.
- Lewnard JJ, Hsiung TH (1987) Temperature effects on catalyst activity in liquid phase methanol process. Stud Surf Sci Catal 38: 141-152.
- Weimer RF, Holley EP, Hsiung TH, Studer DW (1988) Recent research and field modifications for the La Porte LPMeOH process development unit (PDU). 13th Annual Conf on Fuel Sci Conversion, Electric Power Research Institute, Palo Alto, California, USA, pp. 18-19.
- Weimer RF (1985) Slurry reactor technology development for methanol production. NATO Advanced Study Institute: Chemical Reactor Design and Technology, London, pp. 2-12.
- Von Wedel H, Ledakowicz S, Deckwer WD (1988) Kinetics of methanol synthesis in liquid phase. Chem Eng Sci 43(8): 2169-2174.
- Ledakowicz S, Stelmachowski M, Chacuk A (1992) Methanol synthesis in bubble column slurry reactors. Chem Eng Process 31(4): 213-219.
- Park HW, Ha JK, Lee ES (2014) Kinetic mechanism of dimethyl ether production process using syngas from integrated gasification combined cycle power plant. Kor J Chem Eng 31(12): 2130-2135.
- Peng XD, Toseland BA, Underwood RP (1997) A novel mechanism for catalyst deactivation for liquid phase syngas-to-DME reactions. In: Bartholomew C, Fuentes G (Eds.), Stud Surf Sci Catal, Elsevier, Amsterdam, Netherlands, 111: 175-182.
- Wu J, Saito M, Takeuchi M, Watanabe T (2001) The stability of Cu/ZnObased catalysts in methanol synthesis from a CO2-rich feed and from a CO-rich feed. Applied Catalysis A: General 218(1-2): 235-240.
- Roberts GW, Brown DM, Hsiung TH, Lewnard JJ (1993) Deactivation of methanol synthesis catalysts. Ind Eng Chem Res 32(8): 1610-1621.
- Kung HH (1992) Deactivation of methanol synthesis catalysts: A review. Catal Today 11(4): 443-453.
- Dadgar F, Myrstad R, Pfeifer P, Holmen A, Venvik HJ (2017) Catalyst deactivation during one-step dimethyl ether synthesis from synthesis gas. Catal Lett 147(4): 865-879.
- Aguayo AT, Erena J, Sierra I, Olazar M, Bilbao J (2005) Deactivation and regeneration of hybrid catalysts in the single-step synthesis of dimethyl ether (DME) from syngas and CO2. Catalysis Today 106(1-4): 265-270.
- Ordomsky VV, Cai M, Sushkevich V, Moldovan S, Ersen O, et al. (2014) The role of external acid sites of zeolite ZSM-5 in deactivation of hybrid CuZnAl/HZSM-5 catalyst for direct dimethyl ether (DME) synthesis from syngas. Applied Catalysis A General 486: 266-275.
- Garcia-Trenco A, Martinez A (2015) A rational strategy for preparing Cu- ZnO/H-ZSM-5 hybrid catalysts with enhanced stability during the onestep conversion of syngas to dimethyl ether (DME). Applied Catalysis A: General 493: 40-49.
- Jun KW, Lee HS, Roh HS, Park SE (2003) Highly water-enhanced H-ZSM-5 catalysts for dehydration of methanol to dimethyl ether (DME). Bull Kor Chem Soc 24(1): 106-108.
- Schultz H, Wei M (1999) Deactivation and thermal regeneration of zeolite H-ZSM-5 for methanol conversion at low temperature (260 ᵒC - 290 ᵒC). Micro Meso Mat 29: 205-218.
- Schultz H, Wei M (2014) Pools and constraints in methanol conversion to olefins and fuels on zeolite HZSM-5. Top Catal 57(6-9): 683-692.
- Sun JT, Metcalfe IS, Sahibzada M (1999) Deactivation of Cu/ZnO/Al2O3 methanol synthesis catalyst by sintering. Ind Eng Chem Res 38(10): 3863-3872.
- Xu MT, Lunsford JH, Goodman JH, Bhattacharya A (1997) Synthesis of dimethyl ether (DME) from methanol over solid acid catalysts. Applied Catalysis A: General 149(2): 289-301.
- Tartamella T, Lee S (1996) Role of in situ produced methanol on catalyst deactivation in liquid phase methanol synthesis (LPMeOHtm) process. Fuel Sci Tech Int’l 14: 713-727.
- Twigg MV, Spencer MS (2003) Deactivation of copper methanol synthesis catalysts for methanol decomposition, steam reforming, and methanol synthesis. Top Catal 22(3-4): 191-203.
- Twigg MV, Spencer MS (2001) Deactivation of supported copper metal catalysts for hydrogenation reactions. Applied Catalysis A: General 212(1-2): 161-174.
- Quinn R, Mebrahtu T, Dahl TA, Lucrezi FA, Toseland BA (2004) The role of arsine in deactivation of methanol synthesis catalysts. Applied Catalysis A: General 264(1): 103-109.
- Luan YS, Xu HY, Yu CY, Li WZ, Hou SF (2007) In-situ regeneration mechanisms of hybrid catalysts in the one-step synthesis of dimethyl ether from syngas. Catal Lett 115(1-2): 23-26.
- Cheng W (1989) Regeneration of methanol dissociation catalysts. US Patent.
- Ohno Y, Yagi H, Inoue N, Okuyama K, Aoki S (2007) Slurry phase DME direct synthesis Technology-100 tons/day demonstration plant preparation and scale-up study. 8th Natural Gas Conversion Symposium, Natal, Brazil, pp. 27-31.
- Ohno (1999) NKK Technical Review 81: 13-17.
- Ohno Y, Inoue N, Ono M, Shikada T, Hayashi H (2001) Slurry Phase Synthesis and Utilization of Dimethyl Ether. NKK Technical Review 85: 23-28.
- Halder Topse (1996) World Patent Application. WO96/23755.
- Roberts GW, Brown DM, Hsiung TH, Lewnard JJ (1990) Catalyst poisoning during the synthesis of methanol in a slurry reactor. Chem Eng Sci 45(8): 2713-2720.
- Roberts GW, Brown DM, Hsiung TH, Lewnard JJ (1991) In: Bartholomew CH, Butt JB (Eds.), Catalyst Deactivation, Elsevier, Amsterdam, Netherlands, Europe, p. 351.
- Fleisch TH, Basu A, Sills RA (2012) Introduction and advancement of a new clean global fuel: The status of DME development in China and beyond. J Nat Gas Sci Eng 9: 94-107.
- Arcoumanis C, Bae C, Crookes R, Kinoshita E (2008) The potential of dimethyl ether (DME) as an alternative fuel for diesel engines: A review. Fuel 87: 1014-1030.
- Semelsberger TA, Borup RL, Greene HL (2006) Dimethyl Ether (DME) as an alternative fuel. J Power Sources 156(2): 497-511.
- Dybkjar I, Hansen JB (1997) Studies in Surface Science and Catalysis. In: Pontes M, Espinoza RI, Nicolaides CP, Scholtz JH, Scurell MS (Eds.), Nat Gas Conv IV, Elsevier, Amsterdam, Netherlands, Europe, 107: 99-103.
- Rouhi MA (1995) AMOCO, Halder-Topsoe develops dimethyl ether as an alternative fuel. Chem Engng News 73(22): 37-39.
© 2018 Makarand R Gogate. This is an open access article distributed under the terms of the Creative Commons Attribution License , which permits unrestricted use, distribution, and build upon your work non-commercially.
Editor In Chief







.jpg)

























Signup for Newsletter

Quick Links
 Editorial Board Registrations
Editorial Board Registrations Submit your Article
Submit your Article Refer a Friend
Refer a Friend Advertise With Us
Advertise With UsOur Recent Edition

.jpg)



Top Editors




.jpg)

















.bmp)
.jpg)
.png)
.jpg)













.jpg)






.png)

.png)




.png)




Financial Support

Sponsors

Latest e-Books

Latest Video

 a Creative Commons Attribution 4.0 International License. Based on a work at www.crimsonpublishers.com.
Best viewed in
a Creative Commons Attribution 4.0 International License. Based on a work at www.crimsonpublishers.com.
Best viewed in