
- About Us
- Information
-

The Author ensures that the research has been conducted responsibly and ethically with adherence to all relevant regulations. read more..
- For Authors
- For Reviewer
- Manuscript Guidelines
- Membership
- Publication Ethics
-
- Journals
- Reprints
- e-Books
- Videos
- Policies
- Contact Us
 COVID-19
COVID-19
- Submissions



Full Text
Progress in Petrochemical Science
The Direct Dimethyl Ether (DME) Synthesis Process from Syngas: Current Status and Future prospects I. Process Feasibility and Chemical Synergy in LPDMEtm Process
Makarand R Gogate*
Jawaharlal Nehru College of Engineering, India
*Corresponding author: Makarand R Gogate, Jawaharlal Nehru College of Engineering, 259 Samarthnagar, Opp SBI Branch, Aurangabad, India
Submission: May 11, 2018;Published: August 06, 2018

ISSN 2637-8035Volume2 Issue4
Abstract
A novel one-step process for co-production of dimethyl ether (DME) and methanol, in the liquid phase was first conceived by the UA researchers, as an advance over the liquid phase methanol synthesis process (LPMeOHtm). The one-step, direct DME process (LPDMEtm) is based on the application of “dual catalysis”, where 2 functionally different yet compatible catalysts are used as a physical mixture, well-dispersed in the inert liquid phase. Three different reactions, methanol synthesis (via CO and CO2), water-gas shift, and methanol dehydration (to form DME) take place over the 2 catalysts, Cu/ZnO/Al2O3 and typically γ-Al2O3. The favorable thermodynamic and kinetic coupling of methanol dehydration reaction (very rapid and at/near thermodynamic equilibrium) with the methanol synthesis reaction (slower kinetics and highly thermodynamic) leads the beneficial “chemical synergy”. This synergy helps to overcome the limitation on thermodynamic equilibrium conversion, and increases the per-pass syngas conversionand reactor productivity. The catalyst deactivation phenomena in LPDMEtm processes also greatly alleviated compared to methanol alone; the increase in syngas conversion and methyl equivalent productivity (MEP) are sustained over a longer on-stream time.
Here, we review the salient developments in the LPDMEtm process since its inception, first at UA research laboratories and elsewhere including Air Products and Chemicals, Inc. First, we demonstrate the rationale of the LPDMEtm process, and outline briefly the research studies in the two processes, that illustrate the chemical synergy in the LPDMEtm process. This successful example of “cooperative catalysis” can be adapted in principle to many other organic reactions. We then briefly discern the intrinsic kinetics of the LPMeOHtm and LPDMEtm systems, and also shed light of the catalyst deactivation phenomena in these processes. In closing, we outline the reactor design/scale-up and plant operational experience of the 3 commercial technologies, as currently practiced by JFE holdings, BP-AMOCO, and Halder-Topsoe.
keywords: Natural gas; Steam reforming; Coal; Syngas; Methanol; DME, Bi-functional catalysts; Cu/ZnO/Al2O3; γ-Al2O3, Slurry reactors; Bubble column reactors; Chemical synergy; Methyl equivalent productivity (MEP); Intrinsic kinetics; Phase equilibrium; Chemical reaction equilibrium; Catalyst deactivation
Introduction
figure 1: Commercial technologies that are currently available for conversion of syngas conversion to value-added chemicals. The direct DME process is the one covered in this report.

Coal and natural gas as fossil fuels continue to be at the nation’s forefront of energy conversion and power generation processes. In the United States, abundant and plentiful supply of natural gas coupled with its very low costs (~$2MM/BTU and projected to even descend further) has made it an ideal feedstock for conversion to syngas (a mixture of CO and H2, via the highly endotherm steam reforming or auto-thermal steam reforming). Syngas is a very versatile carbon source that is the primary feedstock for further conversion to value-added chemicals (Figure 1). The recent emphasis on biomass (now primarily lignocellulosic woody feedstocks), considered to be a “renewable” and “sustainable” energy source, has made also this an appealing source of our energy needs.
Syngas generation is the first key step in further conversion to methanol and dimethyl ether (DME). The stoichiometric composition of the syngas is a strong function of the type of C source; coal (lignite, peat, or bituminous/subbituminous), natural gas, or biomass, and gasifier type. The low rank carbon sources such as coal (with low H/C ratios) result in a CO-rich syngas (H2/ CO < 1), while the high CV fuels such as natural gas (with high H/C ratios) lead to stoichiometric or balanced syngas, or, a composition which reflects the stoichiometry of methanol synthesis reaction via CO hydrogenation (which requires 2 moles of H2 per mole of CO). The syngas compositions also contain small proportions of CO2 (from total combustion of C) and CH4 (both typically < 5%). The controlling mechanism of methanol synthesis reaction and vapor or liquid phase mode of operation governs the choice of feedstock and the H2/CO ratio.
The introduction of a liquid phase process, in 1975, termed as “liquid phase methanol synthesis process (LPMeOHtm) by Chem Systems, Inc., has been seen as trendsetting in syngas conversion processes [1-3]. In the LPMeOHtm process, a finely powdered methanol catalyst (of the order of 100μm or less) is dispersed or slurried in high-boiling hydrocarbon solvent inert oil). Apart from the high boiling point, other key desirous characteristics of these solvents include a high solubility for syngas components, H2, CO, CO2, and CH4, and a very minimal interaction between the solvent and catalyst.Some of the key advantages of the LPMeOHtm process include better heat transfer characteristics and isothermal operation, use of the CO-rich syngas (from low-cost coal sources), and a very high chemical selectivity to methanol. In the United States, the process feasibility and development studies on the new LPMeOHtm process were initially undertaken by Universities of Akron and Pittsburgh, and Air Products and Chemicals, Inc. (APCI), sponsored by Electric Power Research Institute (based in Palo Alto, Calif.) and Dept. of Energy (a United States Federal agency, with headquarters in Washington, D.C). The UA researchers, since 1985, carried out process development studies in various fundamental and applied aspects, i.e., demonstration of process feasibility, intrinsic kinetics, process chemistry, thermodynamic analysis and development of a software package for combined phase and chemical reaction equilibria for this multiphase and multicomponent system (which enables one to compute the concentrations of dissolved syngas components in the liquid solvent phase, at given reaction conditions), external mass transfer analysis, thermal stability, and scale-up [4-8]. The UA researchers’ also first conceived the direct one-step DME synthesis process, termed as LPDMEtm process [9-12] and later, the DME-to-olefins and DME-to-hydrocarbons processes [13-16], both enhancements over Mobil Oil’s original methanolto- gasoline and methanol-to-olefins process [17-20]. The APCI component has been more focused on catalyst deactivation studies and feasibility/demonstration studies on the pilot scale (5 TPD & 10 TPD scale), of the LPMeOHtm and LPDMEtm processes, at its Alternative Fuels Development Unit (AFDU) in LaPorte, Texas [21- 24]. Later, in 1996 Eastman Chemical Company (based in Kingsport, Tennessee) assumed a major role in the process development and pilot scale/commercial scale operation (in partnership with APCI), and formed a separate corporate entity, the Air Products Liquid Phase Conversion Company, L.P. – with support from U.S. Department of Energy [25,26].
figure 2: A schematic diagram of the LPMeOHtm process in operation at Eastman’s Coal-to-Chemicals complex at Kingsport, Tennessee.

figure 3: A schematic of the commercial bubble column slurry reactor (LPMeOHtm reactor) design

The LPMeOHtm Demonstration Project at the Kingsport site is a $213.7 million cooperative agreement between the U.S. Department of Energy (DOE) and Air Products Liquid Phase Conversion Company, L.P, a partnership between APCI and Eastman Chemical Company. The commercial scale reactor systems at Eastman’s coal-to-chemicals complex, in Kingsport, Tennessee, are based on the bubble column slurry reactor (BCSR) designsand are one of the largest; the reactor main is 7.5ft (or, 2.286m) in diameter and 70ft (21.34m) tall, the design capacity is 260 short tons/day, at nominal conditions of 1000psig (or, 70atm) and 60 oF (or, 315 ᵒC). It is interesting to note that this represents a very significant scale-up from APCI’s prior experience at the AFDU (in LaPorte, TX), where the nominal diameters were 1.5-2ft (0.457-0.609m). The LPMeOHtm technology, in operation at the Eastman’s coal-tochemicals complex, is illustrated in Figure 2. The basic reactor design of the commercial bubble column slurry reactor is given in Figure 3. The new corporate entity, Air Products Liquid Phase Conversion Company, L.P., formed for this purpose (to demonstrate LPMeOHtm/LPDMEtm process on commercial scale) has successfully carried out demonstration runsof these processes at this complex.
Discussion
From LPMEOHTMto LPDMETM – the direct DME synthesis process
Since its discovery in 1975, the LPMeOHtm and LPDMEtm processes have been illustrative examples of how a mature technology on a commercial scale (ICI low temperature methanol synthesis process) can be successfully adapted to a liquid phase operation. The LPMeOHtm process is a highly flexible process that is well-suited to process low-value CO-rich syngas feeds, uses milder reactor design conditions (of temperature and pressure), and a simpler one from a process engineering standpoint. The direct, onestep DME synthesis process is based on the concept and application of so-called “dual catalysis” where 2 functionally different catalysts in a physically admixed form are finely dispersed in a slurry phase reactor system. The 2 functionally different yet compatible catalysts catalyze three parallel reactions: Methanol synthesis (from hydrogenation of CO/CO2), water gas shift, and methanol dehydration to produce DME. The process is based on application of dual catalysis in a single reactor stage, and based on a combination of an equilibrium-limited reaction (methanol synthesis) and an equilibrium unlimited reaction (methanol dehydration). The process chemistry is represented by the following 3 equations:
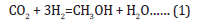
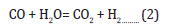
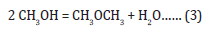
The first two reactions take place over the co-precipitated Cu/ZnO/Al2O3 catalyst and the third one takes place over γ-Al2O3 catalyst. The selective chemical removal of product methanol, from Reaction (1), via its dehydration, via Reaction (3), overcomes the chemical equilibrium barrier on methanol synthesis alone, and improves the per-pass syngas conversion and reactor productivity.
The process chemistry and dynamics of the LPDMEtm process, i.e., synthesis of methanol and DME from CO/CO2/H2 mixtures, is very interesting and can be complicated at times.The chemical synergy in this process was first noted by researchers at Mobil Oil Company [27] and given as comparisons in a series of curve-pairs that relate the overall conversion of syngas (plotted as ordinate) to the proportion of H2 in the feed syngas (plotted as abscissa), each at a specific T and P conditions. In these comparisons, the lower curve represents a methanol synthesis process over a standard Cu/ ZnO/Al2O3 catalyst, with a nominal molar ratio of 60%:25%:15%. The upper curves represent the operation under a DME+ MeOH co-production mode with an identical Cu/ZnO/Al2O3 catalyst for methanol synthesis, but which also has a methanol dehydration component, γ-alumina, incorporated therein (10% by weight). The illustrative comparison is given in Figure 4a-4d. It is clear that the three reactions in the LPDMEtm process chemistry give rise to a very interesting interplay, based on a dual thermodynamic–kinetic coupling, which results in higher syngas conversion and per-pass volumetric reactor productivity, than in the LPMeOHtm mode alone. As stated above, this interplay and thermodynamic-kinetic coupling is often referred to as “chemical synergy”.
figure 4a: A schematic of the commercial bubble column slurry reactor (LPMeOHtm reactor) design

figure 4b: A schematic of the commercial bubble column slurry reactor (LPMeOHtm reactor) design

figure 4c: A schematic of the commercial bubble column slurry reactor (LPMeOHtm reactor) design
figure 4d: A schematic of the commercial bubble column slurry reactor (LPMeOHtm reactor) design

The chemical synergy in LPDMEtm process
As noted above, the chemical synergy in the direct, one-step DME process was perhaps first noted by the researchers’ at Mobil Oil Corporation [27]. The researchers’ at University of Akron and Air Products and Chemicals, Inc., were pioneering in the LPDMEtm studies, including process chemistry, process feasibility, roles of CO/CO2/H2, thermodynamic analysis, and intrinsic kinetics of LPMeOHtm and LPDMEtm systems. We now consider the research portfolios of the University of Akron and Air Products, Inc.
Experimental studies at university of Akron
We first highlight the author’s own experimental studies on the LPDMEtm process, carried out during his doctoral studies at University of Akron. We will next discuss the APCI work on process feasibility and chemical synergy. It is clear from the foregoing discussion that the forward water-gas shift, in which all primary reactants, CO, CO2, H2O, and H2, participate, plays a very crucial role in the overall chemistry.
At the UA research component, the experiments were carried out in a 1-liter stirred autoclave fitted with a 6-blade turbine impeller, within a high-temperature, high-pressure slurry reactor system. The nominal temperature and pressure conditions were 250 oC and 70atm. A feed syngas with nominal composition of H2: CO: CO2: CH4 = 37.4:46.3:8.6:7.7 was used for all experiments. This composition is typical of syngas from Koppers-Totzek or Texaco gasifiers, corresponding to CO-rich syngas, with a H2:CO ratio of 0.8. The nominal feed flowrate was 1 SLPM which corresponded to 2.678mol/h.
The chemical synergy in the LPDMEtm process system was illustrated with 3 different catalyst slurry ratios, as follows:
A. 15g Cu/ZnO/Al2 O3 catalyst (labeled as EPJ-19, UCI/BASF) + 0.5g γ-Al2 O3 catalyst (overall slurry ratio = 4.5%)
B. 80g Cu/ZnO/Al2 O3 catalyst + 5g γ-Al2 O3 catalyst (overall slurry ratio =20%)
C. 150g Cu/ZnO/Al2 O3 catalyst + 10g γ-Al2 O3 catalyst (overall slurry ratio =33%)
The starting volume of the catalyst slurry was 550mL Witco-40 mineral oil (under ambient conditions). The density of Witco-40 oil is a strong function of T and compared to a R.T. value 0.773 g/cm3, its value at 250 oC is only 0.613g/cm3. It is interesting to note that these slurry ratios cover the entire range of practical conditions of interest, from gas-to-liquid mass transfer free to gas-to-liquid mass transfer limited regions. Of course, the chemical synergy and the % increase in MEP productivity is expected to be higher for higher slurry ratios.
Table 1:The chemical synergy in the LPDMEtm process as function of catalyst slurry ratio (at nominal reactor conditions of T=250 0C, P=70atm, syngas flow=2.678mol/h, impeller speed=1500rpm, 1liter stirred autoclave).

The experimental data for the LPMeOHt and LPDMEtm process systems is given in Table 1. At the outset, it is very interesting to note that the chemical synergy “exists” in LPMeOHtm system, even alone. For the three catalyst slurry ratios, 4.5%, 20%, and 33%, the syngas conversion is at 36.9, 45.3, and 48.3%, for LPMeOHtm case. The overall syngas conversion is actually higher than the syngas conversion at chemical equilibrium, 37.5%, at these conditions, T=250 ᵒC, P=70atm, and % H2 in feed syngas=37%. This is a reflection of very facile kinetics of forward water gas shift reaction, which also takes place at or near chemical equilibrium. At these reaction conditions, the K-value for forward WGS reaction is about 1000 times higher than either the CO or CO2 hydrogenation reactions [28,29].
It is worth noting that, from a scientific standpoint, the shift in chemical equilibrium which results in higher syngas conversions and methanol space-time yields, can be thought to work on the basis of “phase equilibrium” or partitioning of methanol in vapor phase and liquid phase, or net condensation of methanol, even in a purely vapor phase gas-solid reaction. There are other reports that validate this working principle of phase partitioning [30]. The experimental observations indicate that methanol exhibits a rather unusual phase behavior around its critical point, 510K and 81atm.
Research portfolio of APCI, Inc.
We now consider the research portfolio of Air Products and Chemicals on the single-step syngas to DME process. Over the past 15 years, Air Products &Chemicals, Inc., with Department of Energy (DOE) sponsorship, has been actively engaged in research and development for LPMeOHtm and LPDMEtm processes, with the overall objective to produce methanol, DME, and other chemicals based on methanol/DME as feedstocks, with the ultimate goal to produce bulk chemicals/fuels/fuel additives from synthesis gas (syngas) generated from coal and natural gas. In APCI research studies, all kinetic experiments on one-step syngas to DME process (LPDMEtm) were carried out in 300cm3 stirred autoclave reactor systems. For the LPDMEtm process, a γ-Al2O3 based methanol dehydration catalyst was used in a physically admixed form, with Cu/ZnO/Al2O3 methanol catalyst, in an 80:20 weight ratio. The experimental conditions used for all experiments were 250 ᵒC, 5.2MPa (or, 52atm), and a gas hourly space velocity of 6,000lit/kg cat.h. Under these conditions, the experiments were free from all internal and external transport gradients, and thus under kinetic control.
A first important element of the APCI one-step DME process research is the influence of H2: CO ratio in the feed syngas on the LPDMEtm reactor productivity and comparison to LPMeOHtm productivity. A new term, called “methanol equivalent productivity”, abbreviated as MEP, was defined for the LPDMEtm process, which was simply defined as the methanol productivity plus 2 times the DME productivity. The comparison of MEP productivity for LPDMEtm and LPMEOHtm processes, as a function of H2: CO ratio in syngas, is illustrated in Figure 5. The chemical synergy is immediately seen from Figure 5. The MEP of LPDMEtm (upper curve) is always greater than that of LPMeOHtm process (lower curve). However, it is clearly seen that the magnitude of the synergy varies with H2:CO ratio. For a H2:CO above 1.5, the percentage increase in MEP is only around 20%. However, this increase becomes 45% at a H2:CO of 1.0 and >90% at a H2:CO of 0.5. In other words, the chemical synergy is the highest at the CO-rich end (highly non-stoichiometric) of the syngas compositions. The LPDMEtm process is also uniquely flexible and can be adapted to IGCC power production combined with oncethrough methanol/DME option.
figure 5: The methanol equivalent productivity (MEP) from LPDMEtm (▲), the MEP from LPMeOHtm (Δ), and the % increase in the MEP from LPMeOHtm to LPDMEtm (•) as a function of H2:CO ratio in the feed syngas.

To further understand the dependence of the chemical synergy on the H2:CO ratio in feed syngas, it is instructive to examine if the system is under kinetic control: The change in MEP was assessed by numerical simulations for 4 independent cases under different regimes of kinetic control. (Figure 6) depicts the MEP for the 4 cases, as follows: (a) the base catalyst system (at km, kd and kw), (b) the system with km increased by a factor of 4 (4 km, kd, kw), (c) the system with kd increased by a factor of 4 (km, 4 kd, kw) and (d) the system with kw increased by a factor of 4 (km, kd, 4 kw). Figure 6 also shows the MEP curve for the system of 3 chemical reactions at chemical equilibrium (solid line). The cases, (●) km, 4kd and 4kw; (○) km, kd, 4kw, appear as 2 curves immediately up top on the (x) km, kd, kw, i.e., the base catalyst system case. In fact, the curve symbols for the base catalyst case and the 4kw case almost overlap. It is clearly seen that quadrupling the rate constant kd and kw, for methanol dehydration reaction and water gas shift reaction, does not exert any appreciable influence at all, on the MEP productivity. The only kinetic rate constant that profoundly influences the MEP productivity is km, the rate constant for methanol synthesis. From the (▲) 4km, kd and kw case, it is seen that increasing the kinetic rate constant for the methanol synthesis reaction raises the MEP productivity from about 40% to 55%.
figure 6: The methanol equivalent productivity (MEP) as a function of H2:CO ratio in the syngas feed. Symbol legend: (x), km, kd, kw, i.e., base catalyst system; (●), km, 4kd, kw; (▲), 4km, kd, kw; (Δ), km, k, 4kw. The solid line over a H2: CO ratio of 0.5-2.0 is represents MEP at equilibrium.

From Figure 6, it is clear that the overall LPDMEtm system is still under “thermodynamic control”. It is thus possible to increase the MEP productivity (and % syngas conversion for LPDMEtm case) if we are able to increase the kinetic rate constants of the three component reactions (with, say, design of more active and selective catalysts that are effective at low temperatures, compared to Cu/ ZnO/Al2 O3 system, which requires 230-270 ᵒC). These cases are denoted by 4km, 4kd, and 4kw, respectively.
Role of the water gas shift reaction
We now make a few brief remarks on one of the long-standing controversial topics in methanol synthesis chemistry, the principal source of C in CH3OH, i.e., CO vs. CO2, OH, and the role of water gas shift reaction. We again point to the interested reader a few very important and pertinent contributions by UA researchers’ to provide unequivocal explanations to this controversial area [4,5,9]. The works of Lee & Parameswaran [5] are the most instructive in this regard. Based on extensive experimental studies and detailed thermodynamic and kinetic analysis of the overall reaction mechanisms, it has been conclusively proven that, under a variety of syngas types (or H2 /CO ratios) – from no-CO2 to no- CO, and balanced (or, stoichiometric, with H2 /CO=2-2.5) to CO-rich, and experimental conditions, the primary dominant pathway for methanol synthesis and the principal source of C in CH3 OH is the hydrogenation of CO2,OH.
From a historical standpoint, the early proposals on the reaction mechanisms and roles of CO/CO2 in the vapor phase methanol process implicitly assumed that CO was the primary source for methanol (perhaps quite naively because CO was in large excess in typical feed syngas streams, compared to CO2 ), for typical process conditions of H2 < 70%, CO+CO2 =25-30%, CO/CO2=15-20%, CH4< 5%, 70-100 bar, and 500-650K. The prevailing (but erroneous) viewpoint of CO hydrogenation was pushed forward further by Klier and his team at Lehigh University [30-34]. However, this viewpoint was countered by the early Russian reports from Rozowski & Kagan [35], who clearly showed that over Cu based catalysts under low temperature conditions, CO2 is the primary source of methanol, i.e., methanol is produced primarily via hydrogenation of carbon dioxide, a reaction which is accompanied by the reverse water gas shift reaction, i.e., hydrogenation of CO2 . More recent isotopic labeling experiments (using labeled reactants, 14C/12C) have provided further evidence for CO2 being the primary source, based on analysis of surface elementary reactions and rate controlling steps [35,36]. This theory is now confirmed by several researchers including Skryzpek [37,38] and other recent works from Korean research groups [39,40].
To further underscore the very important role of water gas shift reaction in the overall LPMeOHtm and LPDMEtm process chemistry, we briefly invoke the kinetic rate models of Graaf et al. [41,42]. We refer to the interested reader to those original works for details of the kinetic model development and kinetic analysis, and some additional details are included in Part 2 of this Series on “Kinetic Studies and Catalyst Deactivation”. Here, we summarize the key findings from their work on a semi-quantitative basis. From the kinetic analysis, it is clear that the primary source of C in CH3 OH is CO2 , i.e., methanol synthesis occurs primarily via CO2 hydrogenation in the liquid phase process (as against vapor phase process, where up to about 30mol %C can be derived from CO; CO2 is still the primary source, however) [43-47]. If one closely examines the kinetic parameters, it is clearly evident that the both pre-exponential factor, 7.21x1017, and apparent activation energy, 215,130J/mol, are very high for the reverse WGS reaction. In fact, the kinetics of the WGS reaction, on a first approximation based on preexponential factors, is faster by about of a factor of 1012compared to CO hydrogenation and 1018 compared to CO2 hydrogenation [48- 52]. This indicates that this reaction plays a very crucial role in the reaction mechanisms in the liquid phase process. Under conditions of low volumetric gas flow rates (or, low WHSV’s), the WGS reaction is even pushed further above the “equilibrium line”: This overshoot is at least 4-times higher for the WGS kinetics in the liquid phase than under corresponding conditions in the vapor phase. The forward WGS reaction, with its very rapid kinetics, is thus critical for the success of the “chemical synergy” phenomena, observed in LPMeOHtm and LPDMEtm processes [53,54].
Summary
The future of DME as an alternative fuel and a chemical intermediate/commodity for targeted end use is very bright. With the right alignment and balance of the political, economic/financial, and environmental “forces”, coupled with the projections of the geopolitical climate, the future use of DME can make impactful and lasting contributions to a nation’s energy and economic security, by virtue of its safe, reliable, and cost-effective supply chain economics. The direct, one-step DME process as described in this paper and the various elements that comprise the current research areas on bench-scale and pilot scale (principally at UA and APCI research in the United States and Halder Topsoe in Denmark) has now formally made the transitional debut at the commercial scale. It is clear that the chemical synergy of the LPDMEtm and LPMeOHtm processes offer cost-effective alternatives to vapor phase modes of operation and provide significant operational and capital savings in plant operations. Dimethyl ether (DME) is being touted for its potential as a clean-burning alternative fuel and a “green” substitute for diesel, as well as a LPG substitute, mainly in Southeast Asia.
References
- Sherwin M, Blum D (1979) Liquid phase methano. Final Report EPRI AF- 1291. Electric Power Research Institute, Palo Alto, California, USA.
- Espino RL, Pletzel TS (1977) Methanol production in a paraffinic medium. US Patent 4,031,123.
- Sherwin M, Blum D (1975) Methanol synthesis in a three-phase slurry reactor. American Chemical Society Fuel Division Reprints 20: 146-151.
- Lee S (1990) Methanol Synthesis Technology. CRC Press, Boca Raton, Florida, USA.
- Lee S, Parameswaran V (1990) Reaction mechanism in liquid- phase methanol synthesis process. Electric Power Research Institute, California, USA, pp. 1-206.
- Parameswaran V, Gogate M, Lee BG, Lee S (1991) Mass transfer in the liquid phase methanol synthesis process. Fuel Sci Tech Int’l 9: 695-744.
- Sawant AV, Lee S, Foos A (1988) Crystal size growth in the liquid phase methanol synthesis catalyst. Fuel Sci Tech Int’l 6: 367-379.
- Sawant AV, Ko MK, Parameswaran V, Lee S, Kulik CJ (1987) In-situ reduction of a methanol synthesis catalyst in a three-phase slurry reactor. Fuel Sci Tech Int’l 5: 77-88.
- Lee S, Sardesai A (2005) Liquid phase methanol and dimethyl ether synthesis from syngas. Top Catal 32(3-4): 197-207.
- Sardesai A, Lee S (1998) Liquid Phase Dimethyl Ether (DME) Process: A Review. Rev Proc Chem Eng 1: 141-178.
- Gogate M (1992) A novel single-step dimethyl ether (DME) synthesis process from syngas. PhD Dissertation, University of Akron, Akron, Ohio, USA.
- Lee S, Gogate M, Kulik CJ (1992) A novel single-step dimethyl ether (DME) synthesis process in a three-phase slurry reactor from CO-rich syngas. Chem Eng Sci 47(13-14): 3769-3776.
- Sardesai A, Tartamella T, Lee S (1999) Performance of ZSM-5 catalyst in the dimethyl ether to olefins process. Petroleum Sci Technol 17(3-4): 273-289.
- Sardesai A, Tartamella T, Lee S (1996) Synthesis of hydrocarbons from dimethyl ether: Selectivities towards light hydrocarbons. Fuel Sci Tech Int’l 14: 703-712.
- Lee S, Gogate M, Fullerton K(1995) Dimethyl Ether to Hydrocarbons/ Gasoline Process. US Patent No. 5,459,166.
- Gogate M, Lee S, Kulik CJ (1995) Methanol-to-Gasoline vs. DME-togasoline II. Process comparison and analysis. Fuel Sci Tech Int’l 13: 1039-1057.
- Meisel SL(1988) Catalysis research bears fruit. Chem Tech 1: 32-37.
- Chang CD, Silvestri AJ (1987) MTG origin, evolution, operation. Chem Tech 10: 624-631.
- Brake LD (1986) Preparation of dimethyl ether from catalytic dehydration of methanol. US Patent 4,595,785.
- Dyer PN, Pierantozzi R(1986) Catalyst for selective conversion of synthesis and method of making the catalyst. US Patent 4,619,910.
- Peng XD (2002) Kinetic understanding of the syngas-to-DME reaction system and its implications to process and economics. Topical Report, Prepared for the United States Department of Energy under Contract No.: DOE-FC22-95 PC93052, Air Products and Chemicals, Inc., Allentown, Pennsylvania, USA.
- Peng XD (2002) Development of kinetic models for the liquid phase methanol synthesis (LPMeOHtm) process. Topical Report, Prepared for the United States Department of Energy under Contract No.: DE-FC22-94 PC93052, Air Products and Chemicals, Inc. Allentown, Pennsylvania, USA.
- Peng XD, Wang AW, Toseland BA, Tijm PJA (1999) Single-Step Syngas-to- Dimethyl ether processes for optimal productivity, minimal emissions and natural gas-derived syngas. Ind Eng Chem Res 38(11): 4381-4388.
- Brown DM, Bhatt BL, Hsuing TH, Lewnard JJ, Waller FJ (1991) Novel technology for the synthesis of dimethyl ether from syngas. Catal Today 8(3): 279-304.
- Heydorn EC, Diamond BW, Lilly RD (2003) Commercial-scale demonstration of the liquid phase methanol synthesis process (LPMeOHtm). Final Report: (Volume 2), Project Performance and Economics, Air Products Liquid Phase Conversion Company, L.P., Allentown, Pennsylvania, USA.
- DOE/NETL Report, Heydorn EC, Diamond BW (2003) Department of Energy Report, Commercial-scale demonstration of the liquid phase methanol synthesis process (LPMeOHtm). A DOE Assessment, DOE/ NETL-2004/1199, National Energy Technology Laboratory, Pittsburgh, Pennsylvania, USA.
- Zahner JC (1977) Conversion of modified synthesis gas to oxygenated organic chemicals. US Patent 4011275.
- Lee S (1990) Methanol Synthesis Technology. Catalysis Methanol.
- Ko M, Lee S (1987) Multi component physical equilibrium of liquid phase methanol synthesis process. Energy & Fuels 1: 211-216.
- Hansen JB, Joensen F (1991) High conversion of synthesis gas to oxygenate in natural gas conversion. In: Holman A (Ed.), Proceedings of Natural Gas Conversion Symposium, Elsevier Science, USA, pp. 457-467.
- Klier K, Chatikavanij V, Herman RG, Simmons GW (1982) Catalytic synthesis of methanol from CO/H2: IV-The effects of carbon dioxide. J Catal 74(2): 343-360.
- Herman RG, Klier K, Simmons GW, Finn BP, Bulko JB, et al. (1979) Catalytic synthesis of methanol from CO/H2: I-Phase composition, electronic properties, and activities of Cu/ZnO/M2O3 catalysts. J Catal 56(3): 407-429.
- Bulko JB, Herman RG, Klier K, Simmons GW (1979) Optical properties and electronic interactions of microcrystalline copper/zinc oxide (Cu/ ZnO) catalysts. J Phys Chem 83(24): 3118-3122.
- Mehta S, Simmons GW, Klier K, Herman RG (1979) Catalytic synthesis of methanol from CO/H2: II-TEM, STEM, micro-diffraction, and energy dispersive analysis of Cu/ZnO and Cu/ZnO/Cr2O3 catalysts. J Catal 57(3): 339-360.
- Rozowski A, Kagan B (1976) About the mechanism of methanol synthesis from carbon dioxide and hydrogen 2: Select the reaction mechanism diagram. KinetikaiKataliz 16(5): 1314-1320.
- Chinchen GC, Waugh KC (1986) The chemical state of copper during methanol synthesis. J Catal 97: 280-283.
- Chinchen GC, Denny PJ, Jennings JR, Spencer MS, Waugh KC (1988) Synthesis of methanol: Part 1. Catalysts and Kinetics. Appl Catal 36: 1-65.
- Skrzypek J, Sloczynski J, Ledacowicz S (1994) Methanol Synthesis. Polish Scientific Publishers, Warszawa, Poland.
- Słoczynski J, Grabowski R, Kozłowska A, Lachowka M, Skrzypek J (2001) Methanol synthesis from CO2 and H2 on Cu/ZnO/Al2O3-ZrO2 catalysts: Catalytic activity and adsorption of reactants. Polish J Chem 75(5): 733- 742.
- Lee JS, Lee KH, Lee SY, Kim YG (1993) A comparative study of methanol synthesis from CO2/H2 and CO/H2 over a Cu/ZnO/Al2O3 catalyst. J Catal 144(2): 414-424.
- Lee JS, Moon KI, Lee SH, Lee SY, Kim YG (1995) Modified Cu/ZnO/Al2O3 catalysts for methanol synthesis from CO2/H2 and CO/H2. Catal Lett 34(1-2): 93-99.
- Graaf GH, Winkelman JGM, Stamhuis EJ, Beenackers AACM (1988) Kinetics of three phase methanol synthesis. Chem Eng Sci 43(8): 2161- 2168.
- Ko M (1987) Thermodynamic analysis and mass transfer study of liquid phase methanol synthesis process. PhD Dissertation, University of Akron, Akron, USA.
- Graaf GH, Stamhuis EJ, Beenackers AACM (1988) Kinetics of low pressure methanol synthesis. Chem Eng Sci 43(12): 3185-3195.
- Beenackers AACM, Graaf GH, Joosten GEH (1987) Recent Trends in Chemical Reaction Engineering. In: Kulkarni BD, Mashelkar RA, Sharma MM (Eds.), , Wiley Eastern Ltd, New Delhi, India, pp. 45-70.
- Lee S, Berty JM, Berty, Greene HL, Desirazu S, et al. (1984) Thermodynamics, kinetics, and thermal stability of liquid phase methanol synthesis process. Electric Power Research Institute, EPRI AP- 3825-SR, Proceedings, Ninth Annual EPRI Contractor’s Conference on Coal Liquefaction, Palo Alto, California, USA.
- Lee S, Sawant A, Parameswaran V, Sullivan T (1985) Research into thermodynamics, mass transfer, oil and catalyst degradation in liquid phase methanol process, Electric Power Research Institute, Proceedings, Ninth Annual EPRI Contractor’s Conference on Clean Liquid and Solid Fuels, Special Report EPRI AP-4253-SR, Palo Alto, California, USA.
- Graaf GH, Sijtsema PJJ, Stamhuis EJ, Joosten GEH(1986) Chemical equilibria in methanol synthesis. Chem Eng Sci 41(11): 2883-2890.
- Brown DM, Gottier GN, Upadhye, RS, Bauer JV, Cilen NA, et al. (1984) Modeling of methanol synthesis in the liquid phase. Chemical Reaction Engineering 87: 699-708.
- Klosek J, Brown DM, Mednick RL (1985) Status of the La Porte LPMeOH process demonstration unit (PDU), Proceedings of Tenth Annual EPRI Contractor’s Conference on Clean Liquid and Solid Fuels, Palo Alto, California, USA.
- Weimer RF, Terry DM, Stepanoff P (1987) Laboratory kinetics and mass transfer in the liquid phase methanol process (LPMeOHtm) process, AIChE Fall Annual Meeting, New York, USA.
- Brown DM, Hsiung TH, Rao P, Greene MI (1985) Catalyst activity and life in LPMeOHtm process. Proceeding of the Tenth Annual EPRI Contractor’s Conference on Clean Liquid and Solid Fuels, Electric Power Research Institute, Palo Alto, California, USA.
- Brown DM, Henderson JL, Hsiung, TH, Studer DW (1990) LPMeOH: Beyond LaPorte-Next steps to commercialization. Proceedings of Fifteenth EPRI Annual Conference on Fuel Science Electric Power. Research Institute, Palo Alto, California, USA.
- Mills GA (1993) Status and opportunities for conversion of synthesis gas into liquid fuels. Fuel 73(8): 1243-1279.
© 2018 Makarand R Gogate. This is an open access article distributed under the terms of the Creative Commons Attribution License , which permits unrestricted use, distribution, and build upon your work non-commercially.
Editor In Chief







.jpg)

























Signup for Newsletter

Quick Links
 Editorial Board Registrations
Editorial Board Registrations Submit your Article
Submit your Article Refer a Friend
Refer a Friend Advertise With Us
Advertise With UsOur Recent Edition

.jpg)



Top Editors




.jpg)

















.bmp)
.jpg)
.png)
.jpg)













.jpg)






.png)

.png)




.png)




Financial Support

Sponsors

Latest e-Books

Latest Video

 a Creative Commons Attribution 4.0 International License. Based on a work at www.crimsonpublishers.com.
Best viewed in
a Creative Commons Attribution 4.0 International License. Based on a work at www.crimsonpublishers.com.
Best viewed in