
- About Us
- Information
-

The Author ensures that the research has been conducted responsibly and ethically with adherence to all relevant regulations. read more..
- For Authors
- For Reviewer
- Manuscript Guidelines
- Membership
- Publication Ethics
-
- Journals
- Reprints
- e-Books
- Videos
- Policies
- Contact Us
 COVID-19
COVID-19
- Submissions



Full Text
Modern Applications in Pharmacy & Pharmacology
Development and Assessment of Dispersible Tablets Containing Lomefloxacin HCl
Mukesh Kumar S1*, Aleza Rizvi2, Basant Lal3, Harshit S4, Ratnanjali P5 and Sakshi S6
1Assistant Professor, Department of Pharmaceutics, Hygia Institute of Pharmacy, India
2Professor and Head of Department, Hygia Institute of Pharmacy, India
3Assistant Professor, Department of Pharmaceutics, Ankerite College of Pharmacy, India
4Associate Professor, Department of Pharmaceutics, Hygia Institute of Pharmacy, India
5Research Assistant, Department of Pharmacology, IIT BHU, India
6Assistant Professor, Department of Pharmacology, Hygia Institute of Pharmaceutical Education and Research, India
*Corresponding author: Mukesh Kumar S, Assistant Professor, Department of Pharmaceutics, Hygia Institute of Pharmacy, India
Submission: April 3, 2023;Published: May 26, 2023

ISSN 2637-7756Volume3 Issue3
Abstract
In the current study, an effort was made to formulate FDT of lomefloxacin HCl in an effort to improve patient compliance, offer a rapid start of action, increase solubility, and cover up its unpleasant taste. By complexing lomefloxacin HCl with Hydroxyl Propyl Cyclodextrin (HP-CD) via a solvent evaporation technique, taste masking and solubility were improved. The finished complex was then directly crushed into tablets utilizing various super disintegrants such as sodium starch glycolate, croscarmellose sodium, and polyplasdone XL-10 at concentrations of 1%, 1.5% and 2% while being sweetened with aspartame and lubricated with aerosol. The amount of medication released by FDT increases as the concentration of super disintegrants rises, and formulation F6-which contains 1.5% croscarmellose sodium and is thought to be the best formulation-releases the most medication-up to 100.68% in 45 minutes. Comparing the FDT of formulation (F6) to regular tablets, in vivo experiments demonstrated that the FDT had an excellent bioavailability. The HP-CD complex rapid dissolving tablet may be made utilizing several super disintegrants using the Direct Compression process, and it was discovered that it disintegrates in less than 2 minutes, resulting in a quicker effect and higher patient compliance.
Keywords: Super disintegrants; Lomefloxacin HCl; Croscarmellose sodium
Introduction
Improvements to API’s physicochemical and biological characteristics, as well as in vivo performance, offer a possible new option in choosing the best solid forms for developing pharmaceutical products [1,2]. Pharmaceutical firms are working to develop new drug dosage forms for current pharmaceuticals with increased safety and efficacy along with reduced dosing frequency and the manufacturing of more cost-effective dosage forms because the development cost of a new drug molecule is quite expensive. Due to its many benefits and higher patient compliance than many other routes, oral routes are still the recommended method of administration for the majority of therapeutic drugs used to elicit systemic effects. The majority of currently available medication delivery devices are tablets and hard gelatin capsules. Unfortunately, numerous patient populations, including the elderly, kids, and patients who are delayed intellectually, uncooperative, queasy, or on liquid restriction diets, have trouble swallowing these dose forms. Also, individuals who are on the go or have limited access to water are also impacted..
Pharmaceutical technologists have created a unique oral dose form called Fast Dissolving Tablets (FDTs) to address these medical demands. FDTs dissolve quickly in saliva, often in a matter of seconds, without the need for water. Drug absorption, solubility, clinical effect onset, and bioavailability may all be much higher than those seen with traditional dose forms [3]. Orally delivered medications’ bitter taste frequently discourages patients from taking their medications, especially youngsters and the elderly. Regrettably, the majority of medications have a naturally bitter taste that might cause tongue or throat burning. Particularly, a bitter taste can reduce patient compliance, which lowers the efficacy of medication. The use of tastes or sweeteners to obtain an acceptable palatability is restricted and may not be effective enough to cover up the taste of medications, necessitating the employment of technical techniques. There are several methods for masking tastes, including the use of ion exchange resins, inclusion complexes with cyclodextrins, viscosity adjustments and melt granulation. A bactericidal fluoroquinolone drug called lomefloxacin HCl has action against a variety of gram-negative and gram-positive pathogens. The bacterial enzymes DNA gyrase and topoisomerase IV, which are essential for the transcription and replication of bacterial DNA, are interfered with by lomefloxacin, which has a bactericidal effect. In the current study, an effort was made to create a fast-acting Lomefloxacin HCl tablet in an effort to improve patient compliance. This tablet will have a quick onset of action, increase solubility, and have its bitter flavour covered. By complexing lomefloxacin HCl with hydroxyl propyl cyclodextrin (HP-CD) in a 1:1 molar ratio by solvent evaporation technique, taste masking and solubility were improved. These complexes were directly crushed into tablets utilizing several super-disintegrants.
Material and Methods
Material
Lomefloxacin HCl was obtained from laboratory of Hygia Institute of Pharmaceutical Education and Research, Lucknow, India. Hydroxy Propyl-β-Cyclodextrin was obtained as a gift sample from Patel Chem Specialties P. Ltd. India. Microcrystalline cellulose, Crospovidone, Croscarmellose sodium, Sodium starch glycolate, were obtained as gift sample from Patel Chem Specialties P. Ltd. India. Aspartame were obtained as gift sample from Dr. Reddy Pvt. Ltd., Hyderabad (India). All other excipients used were of analytical grade.
Methods
Preparation of inclusion complexes: The drug and Hydroxy Propyl-Cyclodextrin (HPBCD) were dissolved in methanol with constant stirring to create an inclusion complex containing lomefloxacin HCl and HPBCD in a 1:1 molar ratio. To create dry granules, the resultant solvent was entirely evaporated at 40-45 °C while being stirred continuously [4].
Formulation of fast dissolving tablet containing a complex of lomefloxacin HCl with HP-β-cyclodextrin: By employing Lomefloxacin HCl and direct compression, fast-dissolving tablets had been manufactured. The HP-CD inclusion complex was created using the solid dispersion/co-evaporated dispersion technique. The drug’s overall identification and micromeritic analysis are displayed in Table 1. Table 2 illustrates the formula, which utilized a range of super disintegrants and other excipients. With the use of a mortar and pestle, an identical amount of the medicine HPCD was taken, combined with super disintegrant and immediately compressible diluents, and then Aspartame was added as a sweetener and Aerosol as a lubricant. A Rimek tablet press machine was then used to compress the mixture. The tablet’s 1000mg total weight was maintained [5-9].
Table 1:Identification and micromeritic study of drug.

Table 2:Composition of fast dissolving tablets of lomefloxacin HCL.

Identification of pure drug
By using infrared absorption spectroscopy, lomefloxacin HCl was identified.
Melting point determination:Melting point of Lomefloxacin HCl was determined by open capillary Method.
Drug-excipient compatibility studies Fourier-transform infrared spectrophotometry:Using the use of a Fourier Transform Infrared Spectrophotometer, the KBr technique was used to record the infrared spectra of Lomefloxacin HCl, HP-CD, and its complexes. The potassium bromide disc technique was used in the current investigation. Dry powdered potassium bromide was thoroughly combined with the powdered sample. Next, using specialized dies and great pressure, this mixture was crushed into a clear disc. The IR spectrometer was used to record the spectrums of this disc. The resolution was 1cm, while the scanning range was 450-4000cm.
Pre-compression parameters
a. Angle of repose: A fixed funnel approach was used to calculate
the angle of repose. A funnel that can be elevated vertically
to achieve a maximum cone height (h) was used to pour the
mixture through. The heap’s radius (r) has been evaluated, and
an equation was used to determine the angle of repose.
Θ=tan-1 h/r Where,
θ is angle of repose,
h is height of pile and r is the radius of the base pile.
b. Bulk density: Pouring the mixture into a graduated cylinder
and determining the bulk volume provided the apparent bulk
density and weight (M) as it is δb=M/Vb .
c. Tapped density: A certain amount of time was spent tapping
the measuring cylinder with the known mass of the mix.
the cylinder’s minimum volume (Vt) as well as the blend’s
measured weight (M). The equation was used to compute the
tapped density (t).
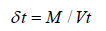
Carr’s compressibility index
Compressibility is the most straightforward technique to quantify a powder’s ability to flow freely. Compressibility is calculated as follows to give an indicator of how easily a material may be made to flow:
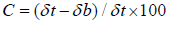
Hausner’s ratio
The formula for calculating Hausner’s ratio, which measures
how easily powder flows, is as follows:
Hausner’s ratio=δt/δb
Where, δt is tapped density and δb is bulk density.
Post compression parameters
Weight variation test: [6] A computerised weighing scale was used to calculate the individual and combined weight of the 20 tablets. The total weight was used to calculate the average weight of one pill.
Hardness test:[6] Six pills were chosen at random, and their hardness was assessed. Using a Monsanto hardness tester, the tablets’ hardness was assessed. Calculated values for the mean and standard deviation are provided in kg/cm2.
Friability:[6] The Roche Fribilator was used to test the
friability of six tablets from each batch, and it was operated for four
minutes at a speed of 25 revolutions per minute. The tablets were
removed, dusted, reweighed, and the percentage of friability was
determined.
%Friability=(Loss in weight/Initial weight)x100
In Vitro Dissolution Studies
Utilizing the tablet dissolution test device USP XXIII at 75rpm and phosphate buffer pH 6.4 as the dissolution medium kept at 37 °C–1 °C, in vitro release studies were conducted. At different time intervals, samples were taken out, diluted, and measured using a UV spectrophotometer at 281nm. The development of in vitro dissolution tests has two goals: (1) to demonstrate that the release of the drug from the tablet is as close to 100% as possible; and (2) to demonstrate that the rate of drug release is constant batch to batch and is identical to the release rate from those batches that have been shown to be bioavailable and clinically effective. The following method was used to calculate the in vitro dissolution rate for each formulation throughout the investigation..
The following list of dissolution-related parameters that the
current work evaluates:
a) Drug release
b) The overall medication release %
c) The overall medication retention rate.
Result and Discussion
Compatibility studies Fourier-transform infrared spectrophotometry studies
Both of the distinctive bands were visible in all spectra, according to infrared measurements. Nevertheless, there were no additional bands or variations in the distinctive peaks.
Pre-compression parameters
To ensure consistency of tablet weight, granules suitable for compression containing medication and diverse excipients were exposed to pre-compression parameters (Micromeritic characteristics). Angles of repose for all formulations were measured and the results were tabulated in Table 3. The values ranged from 230.43’ to 280.76’. Direct compression formulations had poor flow properties, but wet granulation formulations had high flow properties. Table 3 displays the blend’s Loose Bulk Density (LBD) and tapped bulk density (TBD). For all of the formulations combined, the loose bulk density and tapped bulk density ranged from 0.52gm/cm3 to 0.62gm/cm3 and 0.64gm/cm3 to 0.69gm/cm3, respectively. The formulation’s results for Hausner’s ratio were found to fall between 1.108 and 1.211. The Hausner’s ratio was within the acceptable range in all formulations, as indicated in Table 3, indicating that the granules had satisfactory flow characteristics. For all of the formulations combined, the findings of Carr’s consolidation index or compressibility index (%) ranged from 8.99 to 16.23. Table 3 contains the findings for each formulation.
Table 3:Micromeritic study of granules.

Post-compression parameters
The manufactured pills were evaluated in accordance with numerous official standards and other criteria. The following tests were run: hardness, friability, weight fluctuation, disintegration time, in vivo taste, and disintegration. The tablet’s hardness was discovered to be between 4.89kg/cm2 and 6.92kg/cm2, respectively. Table 4 lists the results of the mean hardness test. The formulation’s overall friability ranged from 0.201 to 0.243. In all intended formulations, the acquired findings were discovered to be well within the permitted limit (1%). Table 4 presents the outcomes. All formulations underwent the content uniformity test and the results are listed in Table 4. Table 4 displays the weight variation for each formulation. The average percentage weight variation was determined to be within the Pharmacopoeial limitations of 10%, meaning that all of the pills passed the weight variation test. The findings obtained ranged from 998.9mg to 1001.0mg.
Table 4:Various evaluation parameter of formulations of lomefloxacin HCl (all quantites are taken in mg/ tablet).

In vitro Dissolution Studies
Table 4 presents the outcomes. All formulations underwent the content uniformity test and the results are listed in Table 4. Table 4 displays the weight variation for each formulation. The average percentage weight variation was determined to be within the Pharmacopoeial limitations of 10%, meaning that all of the pills passed the weight variation test. The findings obtained ranged from 998.9mg to 1001.0mg. Using the tablet dissolution tester USP Type 2, in vitro dissolution studies were conducted on all nine formulations. At various time intervals, the samples were taken out and subjected to 281nm analysis. On the basis of the average quantity of Lomefloxacin HCl contained in the relevant tablet, cumulative drug release was computed. Figure 1 & 2 displays the cumulative% drug release vs. time graphs and the in vitro drug release data for the formulations L1 to L3, L4 to L6 and L7 to L9.
Figure 1:Dissolution profile of lomefloxacin HCL complex of various formulations (L1-L6).

Figure 2:Dissolution profile of lomefloxacin HCL complex of various formulations (L7-L9).

It was discovered that the dissolving rate increased linearly with super-disintegrant concentration. At the end of 45 minutes, the drug release was 65.67%, 96.49%, and 97.59% for Formulations L1, L2, and L3, which included Sodium starch glycolate in increasing percentages from 1% w/w to 2% w/w. At the end of 45 minutes, the drug release was 95.89%, 98.19% and 100.54%, respectively, for Formulations L4, L5 and L6, which included increasing amounts of Croscarmellose sodium from 1% w/w to 2% w/w. At the end of 45 minutes, the drug release for Formulations L7, L8 and L9, which included Polyplasdone XL-10 at increasing concentrations of 1% weight/weight to 2% weight/weight, was 96.72%, 97.74% and 99.54%, respectively. By 45 minutes, the drug release was nearly 100% in all formulations. It was necessary to assess the relative effectiveness of several super-disintegrants to increase the pace at which pills dissolve. Polyplasdone XL-10 > Sodium starch glycolate > Croscarmellose sodium (Table 5).
Table 5:Cumulative percent drug release profiles of formulations of lomefloxacin HCl containing different Disintegrants.

Conclusion
The fast-dissolving tablet with hydroxyl propyl beta cyclodextrin (HP-CD) complex can be made using a variety of super-disintegrants, such as croscarmellose sodium, Crospovidone and sodium starch glycolate, and was found to dissolve in less than two minutes, providing a quicker effect and improving patient compliance, according to the results. The prepared tablets demonstrated compliance for a number of physiochemical criteria, including tablet size, hardness, friability, weight fluctuation, homogeneity of content, and disintegration. According to in vitro tests, formulation L6 demonstrated the largest amount of drug release. The formulation F6, which releases up to 100.68% in 45 minutes and contains 2% croscarmellose sodium, can be inferred from the above. Comparing the FDDT of formulation (L- 6) to traditional tablets, in-vivo experiments demonstrated that the former had a good bioavailability. According to the results of the stability investigations, formulations L6 were stable at 40 ˚C/75% RH and the assessed parameters did not significantly change.
References
- Schultheiss N, Newman A (2009) Pharmaceutical cocrystals and their physicochemical properties. Cryst Growth Des 9(6): 2950-2967.
- Jung MS, Kim JS, Kim MS (2010) Bioavailability of indomethacin-saccharin cocrystals. J Pharm Pharmacol 62(11): 1560-1568.
- Hirani JJ, Rathod DA, Vadalia KR (2009) Orally disintegrating tablets: A review. Tropical Journal of Pharmaceutical Research 8(2): 161-172.
- Maski N, Arulkumaran GK, Ghode P, Randive S, Pal R, et al. (2009) Studies on the preparation, characterization and Solubility of β-cyclodextrin-diacerein inclusion complexes. Int J Pharmacy and Pharm Sci 1(2): 121-135.
- Subramanyam CVS (2001) Textbook of physical pharmaceutics, (2nd edn), Vallabh Prakashan Publisher, Delhi, India, pp. 210-228.
- Gohel MC, Bansal G, Bhatt N (2005) Formulation and evaluation of orodispersible taste masked tablets of famotidine. Pharma Biol World 3: 75-80.
- Shoukri RA, Ahmed IS, Shamma RN (2009) In vitro and in vivo evaluation of nimesulide lyophilized orally disintegrating tablets. Eur J Pharm Biopharm 73(1): 162-171.
- McClure N (1997) Stability studies in overview of ICH guidelines for drug products. Matrix Pharmaceutical Inc.
- Shanmugapandiyan P, Selvaraj B, Malarvizhi P, Udayakumar T (2011) Design and evaluation of fast dispersible aceclofenac tablets. Int J Pharm & Ind Res 1(3): 214-218.
© 2023 Mukesh Kumar S. This is an open access article distributed under the terms of the Creative Commons Attribution License , which permits unrestricted use, distribution, and build upon your work non-commercially.
Editor In Chief







.jpg)

























Signup for Newsletter

Quick Links
 Editorial Board Registrations
Editorial Board Registrations Submit your Article
Submit your Article Refer a Friend
Refer a Friend Advertise With Us
Advertise With UsOur Recent Edition

.jpg)



Top Editors




.jpg)

















.bmp)
.jpg)
.png)
.jpg)













.jpg)






.png)

.png)




.png)




Financial Support

Sponsors

Latest e-Books

Latest Video

 a Creative Commons Attribution 4.0 International License. Based on a work at www.crimsonpublishers.com.
Best viewed in
a Creative Commons Attribution 4.0 International License. Based on a work at www.crimsonpublishers.com.
Best viewed in