
- About Us
- Information
-

The Author ensures that the research has been conducted responsibly and ethically with adherence to all relevant regulations. read more..
- For Authors
- For Reviewer
- Manuscript Guidelines
- Membership
- Publication Ethics
-
- Journals
- Reprints
- e-Books
- Videos
- Policies
- Contact Us
 COVID-19
COVID-19
- Submissions



Full Text
Evolutions in Mechanical Engineering
Equilibrium Molecular Dynamical Simulation of Phase Change Material for Energy Storage
Xin Xiao1* and Hongwei Jia2
1 School of Chemical and Process Engineering, UK
2 College of Environmental Science and Engineering, China
*Corresponding author:Xin Xiao, School of Chemical and Process Engineering, Leeds LS2 9JT, UK
Submission: October 24, 2018;Published: November 16, 2018

ISSN 2640-9690 Volume2 Issue1
Abstract
Latent heat thermal energy storage attracts more and more attraction recently. Molecular Dynamical (MD) simulation can be applied to investigate the heat and mass transfer mechanism of phase change materials (PCMs) from the micro point of view, especially for nano-encapsulated and nanoparticle-enhanced PCMs. The paper briefly summaries the status of MD simulations used in the energy storage systems, and the future research topics are pointed out finally.
Keywords: Energy storage; Molecular dynamics simulation; Thermo-physical properties
Introduction
Nanoparticles can be used to keep and increase the thermosphysical properties of the phase change materials (PCMs), such as specific heat capacity. The development and application of nanocomposite is still challenging as the heat and mass transfer mechanism of nanocomposite should be revealed range from molecular scale to macro-scale. Traditional experimental methods show difficulties in predicting macroscopic material properties strongly affected by nanoscale mechanisms. Molecular dynamics (MD) simulation can effectively estimate the materials properties, and the characterization can be helpful for the material design and performance optimization of thermal energy storage and transport PCM. In this study, MD simulations about thermal energy storage have been shortly reviewed, and the method to predict thermal diffusivity and specific heat capacity were summarized accordingly.
Molecular Dynamic Simulation
Models and thermo-physical properties prediction
Figure 1:Typical structures of molecular dynamical simulation. (a) nano-encapsulated PCM fabricated by n-octadecane as core material and SiO2 as shell material, n-nonadecane enhanced with Al nanoparticle [1] (b) domain with Al2O3 and solar salt [2] (c) domain with hydrated 13X zeolite and 76 Na cations [3].

COMPASS [1] and LAMMPS [2] were always used to perform the calculations of molecular potentials. Both NPT (constant pressure and temperature) and NVT (constant pressure and temperature) can be used to equilibrate the system [1]. Figure 1 shows typical structures of MD simulations. Alkane based nano-encapsulated and nanoparticle-enhanced PCM are modeled in Figure 1a. Figure 1b is the domain with Al2O3 and solar salt, and (c) is the domain with hydrated 13X zeolite and 76 Na cations. During the MD simulation, the self-diffusion coefficient (diffusivity) and specific heat capacity of the material can be calculated from Eqs. (1) and (2), respectively [1].
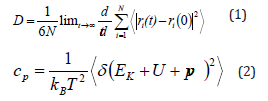
Where N represents the number of atoms, t is the simulation time, and ri is the position vector of the particle i. kB is the Boltzmann constant, T is temperature, Ek is kinetic energy, U is potential energy, p is pressure, and V is volume.
The research of Rao et al. [1] also indicated that it is indispensable to increase the ratio of core material of nanoencapsulated PCM, so as to enhance the mass transfer of nanofluids and heat transport capability. The thickness of the shell material should be simultaneously optimized for the preparation and application of nano-encapsulated PCM. It was found that the mobility of nanoparticle-enhanced PCM decreases with the increase of the diameter of nanoparticles. And the downward trend is more obvious after the nnonadecane melted [3].
Many experiments results show that specific heat capacity increases with the addition of nanoparticle. MD simulation was used to reveal the possible mechanisms for the enhanced specific heat capacity of the nanomaterials. During the investigation of Li et al. [4], an absorbed slip layer of liquid was found at the interface between the nanoparticle and liquid. It was pointed out that the thickness of the layering is about 0.5nm, while the layer moved with the Brownian motion of the nanoparticle. Jo & Banerjee [5] revealed that the potential values of composition and density related to the compressed layer are significantly different from that of the pure salt mixture. It can be seen from Figure 2a & 2b that a compressed layer with high density was formed, which can be attributed to the species concentration of liquid solvent molecules. Figure 2c & 2d show that the addition of nanoparticles creates local concentration gradients, and an obvious compressed layer with large molar ratio of potassium carbonate was found. The partial contribution from the enthalpy of fusion to the formation of the compressed layer caused favorably enhancement of specific heat capacity.
Figure 2:Spatial distribution of density of salt mixtures and molar ratio of potassium to lithium obtained from MD simulations. (a) and (b) represent the density of salt mixtures, (c) and (d) represent molar ratio [5].

Hu et al. [2] deeply investigated the specific heat capacity of nanocomposite with Large-scale Atomic/Molecular Massively Parallel Simulator (LAMMPS) package. The simulation box was made of center location of Al2O3 nanoparticle and random locations of solar salt molecules, as shown in Figure 1b. The well-known Lennard–Jones (L-J) 12–6 potential with long-range Coulombic force was used in the calculation of the interaction between two non-bonded atoms (Eq. (3)), while Berthlot mixing rule was used to calculate the L-J parameters between different atomic species (Eq. (4)).
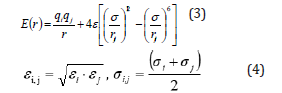
where E(r) and r is the potential of the two atoms and their distance from each other, respectively. q are the charges on atoms, ε is the potential well depth, and σ is the finite distance where the inter-particle potential is zero. Being different from kinetic energy, potential energy can include Van der Waals energy (Evdw), Coulombic energy (Ecoul), long-range k-space energy (Elong) and molecular energy (Emol), as depicted in Eq. (5).
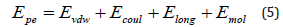
The energy analysis revealed that Coulombic energy significantly increased, inducing the change of the specific heat capacity, as shown in Figure 3.
Figure 3:Potential and interaction energy analysis in the salt/Al2O3 nanofluid system [2].

Extension of Molecular Dynamical Simulation
Furthermore, MD simulation can be combined with other calculation methods, such as Monte Carlo (MC) methods. Fasano et al. [3] combined MC and MD simulations to investigate the heat and mass properties of nanoporous materials for sorption heat storage. A cubic domain with 5.02nm edge (2×2×2 unit cells) was built to compute the isosteric heat of adsorption of the water-13X zeolite and the self-diffusivity of water within the zeolite structure, as shown in Figure 1c. Both the nonbonded and bonded potentials built the force filed. The nonbonded interactions including van der Waals and Coulomb interactions are modelled with Lennard-Jones, while bonded interactions including covalent are modelled with harmonic terms.
Figure 4:(a) Average interaction potential between water-water and water-zeolite molecules (13X zeolite with 76Na, 300K) [3] (b) Water diffusivity of water adsorbed on 13X zeolite (76Na) [3].

MD trajectories was used to obtain the interaction energies, and the energy difference between UWW (water-water) and UWZ (water-water) was considered as the released enthalpy during the adsorption process, which was shown in Figure 4a. Because of the higher kinetic energy of adsorbate molecules according to Arrhenius equation (1), water diffusivity increased with the temperature, as shown in Figure 4b. Ea is the activation energy of the material confining the motion of water molecules, and R is the gas constant.
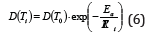
Conclusion
As macroscopic material properties strongly affected by nanoscale mechanisms, MD simulation should be an effective tool to reveal the heat and mass transfer mechanism of the nanomaterials for thermal energy storage. The simulation result showed a compressed layer was formed in the nanomaterial system which attribute to the increase of specific heat capacity.
Furthermore, MD simulation can be combined with other methods, such as First principle, Anharmonic lattice dynamics method (Lattice Boltzmann model), Kinetic Monte Carlo (combination of MC and MD), so as to reveal the heat transfer mechanism extensively. Future works should be progressed as all those simulated methods herein should be helpful for the material design and performance optimization of thermal energy storage and transport PCM.
References
- Rao ZH, Wang SF, Peng, FF (2013) Molecular dynamics simulations of nano-encapsulated and nanoparticle-enhanced thermal energy storage phase change materials. International Journal of Heat and Mass Transfer 66: 575-584.
- Hu YW, He YR, Zhang ZD, Wen DS (2017) Effect of Al2O3 nanoparticle dispersion on the specific heat capacity of a eutectic binary nitrate salt for solar power applications. Energy Conversion and Management 142: 366-373.
- Fasano M, Borri D, Cardellini A, Alberghini M, Morciano M, et al. (2017) Multiscale simulation approach to heat and mass transfer properties of nanostructured materials for sorption heat storage. Energy Procedia 126: 509-516.
- Li L, Zhang YW, Ma HB, Yang M (2008) An investigation of molecular layering at the liquid-solid interface in nanofluids by molecular dynamics simulation. Physics Letters A 372(25): 4541-4544.
- Jo B, Banerjee D (2014) Enhanced specific heat capacity of molten saltbased nanomaterials: Effects of nanoparticle dispersion and solvent material. Acta Materialia 75: 80-91.
© 2018 Xin Xiao. This is an open access article distributed under the terms of the Creative Commons Attribution License , which permits unrestricted use, distribution, and build upon your work non-commercially.
Editor In Chief







.jpg)

























Signup for Newsletter

Quick Links
 Editorial Board Registrations
Editorial Board Registrations Submit your Article
Submit your Article Refer a Friend
Refer a Friend Advertise With Us
Advertise With UsOur Recent Edition

.jpg)



Top Editors




.jpg)

















.bmp)
.jpg)
.png)
.jpg)













.jpg)






.png)

.png)




.png)




Financial Support

Sponsors

Latest e-Books

Latest Video

 a Creative Commons Attribution 4.0 International License. Based on a work at www.crimsonpublishers.com.
Best viewed in
a Creative Commons Attribution 4.0 International License. Based on a work at www.crimsonpublishers.com.
Best viewed in