
- About Us
- Information
-

The Author ensures that the research has been conducted responsibly and ethically with adherence to all relevant regulations. read more..
- For Authors
- For Reviewer
- Manuscript Guidelines
- Membership
- Publication Ethics
-
- Journals
- Reprints
- e-Books
- Videos
- Policies
- Contact Us
 COVID-19
COVID-19
- Submissions



Full Text
COJ Robotics & Artificial Intelligence
Review of Wagner’s Artificial Gene Regulatory Networks Model and its Applications for Understanding Complex Biological Systems
Yifei Wang*
School of Biological Sciences, Georgia Institute of Technology, USA
*Corresponding author: Yifei Wang, School of Biological Sciences, Georgia Institute of Technology, USA
Submission: January 28, 2019;Published: February 26, 2019

ISSN:2832-4463 Volume1 Issue1
Abstract
In the mid of 1990s, Andreas Wagner proposed a gene regulatory network model where the self-development process was explicitly modelled in the system. The many-to-one mapping mechanism of genotype to phenotype in Wagner’s GRN model enables genes to buffer against and even exploit likely variations in the genome. This mechanism is crucial for evolutionary innovations, because genotypes which control gene-gene interactions can change profoundly without affecting phenotypes which represent gene activities or expression concentrations. Wagner’s GRN model motivates research on the evolution of genetic networks and has been successfully employed to study many fundamental evolutionary and ecological questions. In this paper, I will review Wagner’s GRN model and currently available research papers that fall into its framework.
Introduction
Gene regulatory networks control the expression of genes for providing phenotypic traits in living organisms. They play a central role in cells and govern cell differentiation, metabolism, the cell cycle, and signal transduction [1]. Many computational models that aim at capturing essential structure and dynamics of networks have been developed to uncover underlying mechanisms of transcriptional networks in nature [2-7]. Generally, there are two types of network models developed for quantitatively or qualitatively analyzing the evolutionary dynamics of genetic networks [8,9]. The first type of models focuses on modelling a specific network or genetic pathway to quantitatively understand, for example, the segment polarity network in Drosophila [10] the oscillatory network in Escherichia coli. and the cell-cycle network in yeast [11]. These models typical use differential equations and require precise measurements of the concentrations or activities of gene products modelled through biochemical parameters, for example, binding affinities of transcription factors, dissociation constants of the receptors and ligands, or rate constants of enzymes kinetics [8].
However, the quantitative information of parameters used in those models is largely unknown, even for some well-studied experimental systems due to limitations of current biochemical techniques [9,12]. Specially, for many biological networks, we do not have a comprehensive understanding about each circuit in a network interact with whom. Even if such quantitative information is available, it has been difficult to precisely estimate or measure exact strengths of gene-gene interactions. Therefore, due to the lack of quantitative information in studying genetic networks, the second type of models have been used more broadly to discover general principles that emerge from dynamics of genetic networks [8,9]. A recent review of such models can be found in Spirov & Hol-loway [13]. These models typically use general and abstract representations, and, therefore, do not require measurements or estimates of biochemical information in nature systems. One of the most successful computational gene regulatory network (GRN) models was proposed and developed by Wagner [12,14].
The novel feature in Wagner’s GRN model is that it introduces the selection for phenotypic stability, performed as a separate layer of purifying selection1 in addition to the selection for a particular or an optimal phenotype [15,16]. Because of this purifying selection imposed on the population, only individuals that can achieve developmental equilibrium are able to survive during the evolution. The central assumption is that an individual’s phenotype should be able to buffer against genotypic variations. In other words, the selection for phenotypic stability provides a viable simulation for the known natural phenomenon of an individual’s phenotype being expressed relatively stable while its genotype undergoes evolution.
Wagner’s GRN model, initially proposed to describe the evolutionary mechanism of gene duplication [12], has been employed as a popular in silico modelling approach to study epistasis, non-linear interactions between alleles at different loci, and complex genetic interactions for a broad range of fundamental research questions in evolution, ecology and systems biology, for example, gene duplication [14], genetic assimilation and robustness [4,12,17-28], recombination and sexual re-production [16,29-36], phenotypic plasticity [37-41], evolvability [15,42-44], network topology [45], sub-functionalisation [32], incompatibility [46], modularity [47,48], selection strength [49], developmental stability [50,51] and parental E ects [52]. In this paper, I will first review the core implementation of Wagner’s GRN model. Then, I will future review currently available research papers that fall into the framework of Wagner’s GRN model, classifying them into several application areas2 in chronological order.
FOOT NOTE
1Deleterious mutation that impairs the phenotypic stability of the network will be eliminated by the purifying selection.
2Note that the reviewed papers are grouped by their main research focuses. This does not necessarily indicate that they are not relevant to other research topics.
Wagner’s GRN Model
The Wagner’s GRN Model was originally proposed by Wagner [12,14], and developed by Siegal & Bergman [28]. The model typically assumes that different or partially overlapping sets of transcription factors are expressed in different cells or different regions at any given stage of development of an organism [14].
Genotype
In Wagner’s GRN model, the genotype is represented as a network which contains interactions among transcriptional genes. This interaction network encapsulates epigenetic features, such as protein-DNA-binding affinities and transcriptional activation or repression strengths [28,14]. Formally, for each individual network in a finite population M, N cis-regulatory transcription factors are encoded by N*N matrix W (see an example network with four genes in Figure 1. Each element Wi,j (I,J = 1,2,...N ) represents the regulatory effect on the expression of gene i of the product of gene j.
Figure 1:An example of gene regulatory network. (A) Network representation of regulatory interactions among ve genes. Open and lled circles represent genes that are completely activation (+1) or repression (1), respectively. The initial gene expression pattern is s(0) = (+1,+1,+1,+1) This example network is stable as it can reach an equilibrium pattern, which is ( 1, 1, 1, 1) EQ s = + + + + by iterating Equation (1) using the sigmoidal mapping function with a=100. (B) Interaction matrix (W) represents the network in (A). Each element in row i and column j, i.e.,Wi,j (I,J = 1,2,5 ) represents the regulatory effect on the expression of gene i of the product of gene j.

Note that the matrix W is appropriate to be considered as a `genotype’ in the sense that it can be mapped to specific nucleotide sequences in the enhancer regions of the network genes [28]. The network connectivity parameter c determines the proportion of non-zero elements in the network W. A zero entry means there is no interaction between two genes. Through gene interactions, the regulatory effect acts on each gene expression pattern.
Phenotype
In Wagner’s GRN model, the phenotype for a given network W is denoted by a state vector where s(t)=(s1(t),s2(t),....sN(t)) where si(t) represents the expression level of gene (or concentrations of proteins) i at time t. Each value of expression state si(t) is within the interval [-1; +1] that expresses complete repression (-1) and complete activation (+1). Note that for reasons of computational convenience, the expression level or the admissible concentration range for each si(t) can be normalized and restricted to the interval [0; 1]. Note that the model typically assumes that mRNA transcripts and their corresponding protein products are directly proportional in concentration. In other words, there is no posttranscriptional regulation, and, therefore, s(t) can be considered as either transcription or protein concentrations [28,12].
The initial phenotypic state’s (0) is usually assigned random values from [-1, +1] (or [0, 1]), and is fixed throughout individual’s lifetime. This is because the model typically assumes that the initial state is a response to an extracellular signal, such as a growth factor or a specific composition of nutrients in the medium [14]. Therefore, it is assumed that the initial state is determined by the products of one or more ‘upstream’ genes that are not themselves part of the network and it is not regulated by any factors in the network.
Developmental process
In Wagner’s GRN model, it is typically assumed that the expression of transcription factor genes is only in one developmental stage and only in one set of cells (nuclei), for example, a set of nuclei in a part of a Drosophila blastoderm expressing a specific subset of gap genes and pair-rule genes [14]. The basic idea of the developmental process is that individual’s phenotypic state changes over time due to cross regulation and auto-regulation of the expression of member genes by their gene products [14] Figure 2. Formally, for a given gene regulatory network W, the dynamics of s for each gene i is modelled by a set of coupled difference equations:
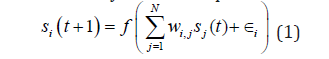
Figure 2:The developmental process in Wagner’s GRN model. Each gene phenotypic state at time t+1,si(t+1) (i=1,2,....N) (diamond boxes on the right), is regulated by the products of the other genes’ phenotypic state at time sj(t)(j=1,2...N) via upstream enhancer factors (square boxes on the left) whose strength and direction of regulation are depicted as different colour saturation levels. The result of additive regulations is then normalised by a mapping function, such as a sigmoidal or a step function. The figure is a modified version of Siegal & Bergman [28].

where f(.) is a sigmoidal function, and εi is a constant which re-acts either a basal transcription rate of gene i or inuences of upstream gene(s) on gene i. In all simulations, unless otherwise specified, I set εi=0 and follow specified, I set i=0 and follow Azevedo et al. [29].
Siegal & Bergman [28] to define f (x) = 2 / (1+ e−ax ) −1 , where a is the activation constant determining the rate of change from complete repression to complete activation. From Figure 3, we can see that when a is large, for example, a=100, f(x) is similar as a step function where f (x) = −1FORx < 0, f (x) = +1FORx > 0 and f (0) = 0 . Therefore, it is earlier to produce extreme values (-1 or +1). The lower values of a, for example, a=1, allow intermediate expression states (Figure 3), but it is difficult to produce extreme phenotypic values. A detailed biological interpretation of the parameter a can be found in Palmer & Feldman [46] where authors summarized that in terms of a metaphorical ‘fitness landscape’, larger values of a correspond to broad-based, sloping hills that are peaked, rather than at, on top, whereas lower values correspond to narrow elevated areas with steep sides and a at top.
Mutation
Generally, there are two kinds of mutations that are usually modelled in the system. The first kind of mutations refers to changes in a given regulatory genotype, W. Specifically, such mutations can 1) cause changes in the existing interactions (non-zero entries in W) by replacing their original interaction strengths with new values drawn from the standard normal distribution N(0, 1) (Figure 4) form new interactions by setting new values drawn from N (0, 1) to zero entries in W or delete the existing interactions by setting their values to be topological mutation rate in. Here, I define the mutation rate in 1) as μ and define the topological mutation rate in 2) as μtop (typically μ ≫μtop). In all simulations, unless otherwise specified, I do not allow any topological mutation, i.e., μtop = 0, and the probability of individual network that acquire k mutations in its non-zero entries is drawn from the Poisson distribution P(x = k) = μk e−u / k ! (k = 0,1,.....,c _ N2 ) . Note that the model does not consider mutations in sequences that code for gene products | mutations that simultaneously affect the interaction for a given gene product with all its target enhancer or promoter regions [28].
Figure 3:The sensitivity of parameter a on changing regulatory responses. At each time step during the developmental stage, the expression level of a gene is determined by a filtering function f (x) = 2 = (1+ eax )1, which normalises the sum of regulatory e ects from other genes. The activation constant a determines the rate of the transition between expression states 1 and +1.

Figure 4:The operators of mutation and recombination in Wagner’s GRN model. Mutation only occurs in nonzero entries in the genotype (see the red box). Recombination occurs by choosing two parental networks (blue and green genotypes) at random to from a transient diploid, which then segregates rows of the matrix to from a single, haploid o spring network.

The second kind of mutations refers to changes in the initial gene expression pattern, s(0). Such mutations have a nongenetic origin that could result, for example, from intracellular noise, from environmental fluctuations, or from disturbances in the activity of genes upstream of the circuit Espinosa-Soto et al. [19] But for reasons of computational convenience, I do not consider any nongenetic mutation in all simulations.
Recombination
In the genotype W, because all entries in the ith(i = 1, 2,....,N ) row represent the promoter or enhancer regions of gene i, we can assume that the individual transcription factor binding sites on those regions are genetically closely linked to one another. Consequently, the recombination will occur only very rarely between them [24]. In contact, different genes in a regulatory circuit are often assumed to be unlinked to one another as they can occur on different chromosomes [32]. In Wagner’s GRN model, the recombination is modelled as a free recombination between circuit genes and neglect recombination within genes (promoters or enhancers) Figure 4. To be more specific, recombination occurs by randomly select two parental networks from the population pool to form a transient diploid. Then for each pair of rows i in the parental networks, one of two rows are chosen with an equal probability to form a single, haploid progeny [32].
Stabilizing selection
In all simulations, network developmental stability is defined as the progression from an arbitrary initial expression state, s (0), to an equilibrium expression state (reaching a fixed phenotypic pattern), by iterating Equation (1) a fixed number of times, dev T. If a given network W can achieve the stability over this developmental time period, it is termed stable, otherwise, it is labelled unstable. Note that this stabilizing selection also refers to as the purifying selection such that those unstable networks will be eliminated. The equilibrium expression state can be reached when the following equation is met:
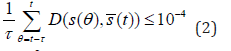
where 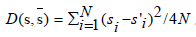 measures the difference
between the gene expression pattern s and s, and s is the average of
the gene expression level over the time interval [t −τ ,t −τ +1,....t)
where _ is a time-constant characteristic for the developmental
process under consideration and will depend on biochemical
parameters, such as the rate of transcription or the time necessary
to export mRNA into the cytoplasm for translation [14].
measures the difference
between the gene expression pattern s and s, and s is the average of
the gene expression level over the time interval [t −τ ,t −τ +1,....t)
where _ is a time-constant characteristic for the developmental
process under consideration and will depend on biochemical
parameters, such as the rate of transcription or the time necessary
to export mRNA into the cytoplasm for translation [14].
Target selection
In Wagner’s GRN model, the target selection refers to the selection for a particular or an optimal phenotype. For networks that can achieve developmental stability (reaching an equilibrium state, sEQ), the phenotypic distance between the equilibrium state and the optimal state D(sEQ,sopt defined in Equation (2), can be used to calculate individual’s fitness. Specifically, there are two measurements that are typically used in the model. The first exponential fitness evaluation function (Figure 5) is defined as in Siegal & Bergman [28], Wagner [14].
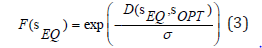
Figure 5:Exponential selection curve for target phenotype. The normalised phenotypic distance x is defined as D(sEQ,Sopt)(see Equation (3)). The fitness output was evaluated under different selection pressure: 𝜎=0:1 (strong selection strength), 𝜎=0:5, 𝜎=1, 𝜎=10 and 𝜎=100 (weak selection strength).

Figure 6:Multiplicative selection curve for target phenotype. The normalised phenotypic distance x is defined as D(sEQ,Sopt)(see Equation (4). The tness output was evaluated under different selection pressure: 𝜎=100 (strong selection strength), 𝜎=10, 𝜎=1, 𝜎=0:1 and 𝜎=0:01 (weak selection strength).

where 𝜎 is the selection pressure that we impose on the population during the evolution. sOPT is usually set to be s (0). Unless otherwise specified, I assign zero fitness to individuals that cannot reach the developmental equilibrium. The second multiplicative fitness evaluation function (Figure 6) is defined as in Draghi & Wagner [42]:
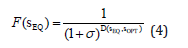
where 𝜎,sOPT ,andsEQ are defined similarly as in Equation (3). Note that for some variants of Wagner’s GRN model, the WIJ are set to be binary values:WIJε{0,1} [43-45]. Then D(sEQ,sopt)can be simply calculated as number of equilibrium gene states that diffier from the optimum.
Evolution process
The reproduction-mutation-selection life cycle is employed in the in-silico evolution. In typical asexual evolution, an individual is chosen at random to reproduce asexually by cloning itself, and then subject to mutation. Similarly, in typical sexual evolution, two individuals are chosen at random to reproduce sexually by recombining two genotypes, and then subject to mutation. Next, the resulting offspring network is exposed to the stabilising selection. Unless otherwise specified in certain evolutionary scenarios, if the offspring network cannot reach an equilibrium state, then it will be wiped out from the population immediately. For the stable offspring network, it is then exposed to the target selection, and can be selected into a new population pool for the next generation based on its fitness calculated by using Equation (3) or Equation (4). In each generation, this process is repeated until the number of M networks are produced.
Applications
Genetic assimilation & robustness
In a classic experiment of Waddington [53], a phenotype of crossveinless wings appeared when Drosophila pupae of a wild Edinburgh strain were exposed to a temperature shock after puparium formation. Waddington then selected those offspring with crossveinless wings and further observed that the crossveinless phenotype continued to appear even when the temperature shock was no longer applied. He referred to this process as genetic assimilation whereby environmentally induced phenotypic variation becomes constitutively produced even if the environmental signal is absent [53,54]. Waddington [53] further envisioned a metaphor for the biological development in which cells, represented by balls, roll downhill through a high-dimensional epigenetic landscape, and described the concept of canalisation (also termed as robustness) as the deepening of valleys (pathways) down the slope, making the developmental outcome less sensitive to perturbations [53-56]. Since Waddington, a large number of studies have focused on uncovering underlying mechanisms by which canalisation can be achieved. However, how canalisation affects the distribution of molecular or genetic variations at different levels of genetic hierarchies or regulatory genes are still unclear [57].
Wagner [12] first employed his GRN model, which explicitly incorporates the self-development along with the evolutionary process, to investigate canalisation in the context of genetic networks and reported that the probability of mutations that cause changes in gene expression patterns can be substantially reduced. He referred to this phenomenon as epigenetic stability, that is, the system of epigenetic interactions may compensate or buffer some of changes that occur as mutations on its lowest levels. Wagner [12] also observed this increased epigenetic stability independently from experiments with variations in network architecture or other model parameters.
Siegal & Bergman [28] developed Wagner’s GRN model, and further showed that the selection for developmental stability is sufficient for canalisation. Specifically, Siegal and Bergman designed evolutionary scenarios where they measured the phenotypic distance of evolved populations in the face of mutation perturbations under different selection pressure for the optimal phenotype. They reported that networks can evolve greater insensitivity to mutation even without the directional selection for this property, that is, the selection for the optimal phenotype is largely absent. They concluded that genetic canalisation, the phenotypic insensitivity to mutation, is an emergent property of complex gene networks.
Masel [25] introduced external noise at individual’s developmental stage, and further reported that the selection for developmental stability is also sufficient for genetic assimilation. Specifically, the modelled noise served as environmental perturbations similarly to the temperature shock as described in the experiment of Waddington [53] can consequently affect the phenotype-genotype mapping. Masel then measured the phenotypic diversity in the presence of noise to access the evolution of genetic assimilation.
In addition to the phenomenon observed by Siegal & Bergman [28], Masel concluded that the results support the utility of Waddington’s canalisation as an explanation for genetic assimilation.
Huerta-Sanchez & Durrett [20] re-examined previous work of Wagner [12] and Siegal & Bergman [28] and proposed a mathematical framework to investigate a simplified version of Wagner’s GRN model in more detail. Huerta-Sanchez and Durrett showed that the qualitative observation that systems evolve to be robust, is itself a robust conclusion, given that the population size is succulently large. They further explained that robust systems by definition of the model are insensitive to mutation and hence have a large mount of viable offspring. Therefore, the evolution of robustness is simply the selection for greater reproduction success.
Ciliberti et al. [8] studied how robustness varies in networks with different architectures. They showed that robustness to mutations and noise are positively correlated. Here the noise was modelled as perturbations to initial gene expression patterns, which is different from the noise introduced at the developmental stage as in Masel [25] Moreover, Ciliberti et al. [8] showed that highly robust networks can be reached from networks with lower robustness through gradual and neutral evolution in one large metagraph of network architectures. In a similar study [8], the same authors further concluded that the robustness emerged from the connected metagraph can simulate a long-term innovation in gene expression patterns.
Kimbrell & Holt [21] studied the canalisation in the source-sink evolution. Here the sink was modelled as a low-quality habitat where populations cannot persist without recurrent immigration from a source population, whereas the source was modelled as a highquality habitat. They showed that the probability of adaptation to the novel habitat decreases when canalisation increases. However, by introducing the noise to gene initial expression patterns as in Ciliberti et al. [8], Kimbrell and Holt found that the noise can facilitate adaptation to novel habitats.
Martin & Wagner [24] investigated how multinationality affects the network robustness to mutations and noise. The multinationality was modelled as different pairs of gene initial and equilibrium expression patterns. They showed that the number of network architectures decreases dramatically as the result of the increased additional functions that they are required to carry out. Giving that the relationship between the robustness of one function and that of other functions to mutations and noise is largely absent, Martin and Wagner concluded that the robustness trade-offs of multiple stable phenotypes generally do not arise in such systems.
Leclerc [23] argued that the common measurement for robustness as used previously in Wagner & Bergman [14,28] may not be appropriated as the measurement inadvertently discounts costs of network complexity. By taking costs of complexity into account, Leclerc showed that a higher robustness could be observed in sparsely connected networks with parsimonious architectures. Moreover, the author showed that selection will favor sparse networks if the network architecture is free to evolve.
Espinosa-Soto et al. [19] introduced non-genetic perturbations and studied the relationship between a phenotype’s mutational robustness and population’s potential to generate novel phenotypic variations. Here, the non-genetic perturbations referred to both perturbations from environmental factors such as temperatures, diets or biotic iterations as modelled in Masel [25] as well as perturbations from an organism’s internal factors such as activity changes in gene initial expression patterns as modelled in Ciliberti et al. [8] and Kimbrell & Holt [21]. Espinosa-Soto et al. [19] found that the phenotypic robustness facilitates variability in response to non-genetic perturbations, but not in response to mutations.
Le Cun & Pakdaman [22] reviewed previous work using Wagner’s GRN model, and derived new observations of emergent properties with respect to the robustness in the system. They showed that selection for a specific (target) phenotype also benefits to increase the probability of stabilizing alternative phenotypes revealed under stress. Le Cun & Pakdaman [22] further showed that a generalized canalisation in the system can drive population towards robustness in the presence of perturbations, for example, gene deletion, loss of 8 interactions and mutations in regulation activities.
Shin & MacCarthy [27] investigated how robustness and sensitivity become distributed in a host-parasite model of antagonistic co-evolution. Here parasites were modelled on species such as cuckoos where mimicry of the host phenotype confers a higher fitness to the parasite but a lower fitness to the host. They found that sensitivity sites are broadly distributed throughout the network and continually relocate. Shin & MacCarthy [27] referred to this phenomenon as `Whack-A-Mole’inspired by a popular fun park game.
Espinosa-Soto [18] employed Wagner’s GRN model and investigated how selection for network stability affects the evolution of robustness. Espinosa-Soto showed that stabilizing selection on different phenotypic properties can increase mutational robustness.
Runneburger & Le Rouzic [26] studied the conditions for canalisation to emerge in the context of Wagner’s GRN model. Runneburger & Le Rouzic [26] confirmed that most of parameters used in Wagner’s GRN model have a less effect on the evolution of genetic canalisation. They showed that the selection for phenotypic optima can have higher canalization levels than the selection for intermediate expression levels.
Recombination & sexual reproduction
Recombination is ubiquitous in multicellular plants, animals and even fungi. But it is still unclear how evolutionary dynamics, such as sexual reproduction, contribute to the stability of inheritance. All sexual systems exhibit recombination, the reshuffling of parental genetic information which generates novel, heritable gene combinations [58-61]. However, sexual reproduction is also considered to be very costly since it may damage well-adapted lineages and produces fewer offspring. Consequently, why sexual reproduction can be maintained? For decades, researchers have made tremendous efforts and proposed numerous possible theories to explain and uncover the mystery of sex and recombination [34,58,61-64]. Two classic benefits of sex and recombination, though still controversial, are [33,60,65-68]: 1) purging deleterious mutations more efficiently, and 2) creating novel gene combinations. However, although many observed phenomena, such as improving robustness and facilitating evolutionary adaptation, can be attributed to sexual reproduction, the underlying evolutionary mechanism is still poorly understood [34].
Azevedo et al. [29] first employed Wagner’s GRN model to study the maintenance of sexual reproduction in the context of genetic networks. They showed that sexual populations can evolve a higher robustness than asexual populations. Moreover, they further observed that synergistic (negative) epistasis can evolve from sexual populations as a by-product of stabilizing selection imposed in the system, whereas antagonistic (positive) epistasis evolves from asexual populations. Azevedo et al. [29] concluded that sexual reproduction evolves genetic properties that favour its own maintenance.
MacCarthy & Bergman [32] pointed out that the study conducted by Azevedo et al. [29] may not explicitly examine whether sexual populations can outcompete asexual populations under the condition of synergistic epistasis. Specifically, they studied conditions whereby asexual reproduction could nonetheless be favoured by allowing the spontaneous emergence of epistasis in the evolution and introducing a modifier locus that explicitly alters the recombination rate. They found that the fixation time of the asexual mode only has a significant correlation between the level of antagonistic epistasis, but not that of synergistic epistasis. MacCarthy & Bergman [32] highlighted the deterministic mutation hypothesis may not be a plausible explanation for the maintenance of sexual reproduction.
Martin & Wagner [33] focused on effects of recombination in the context of genetic networks. They showed that recombination has much weaker effects than point mutations. Moreover, they demonstrated that recombination reduces the genetic load and also increases dramatically the genetic diversity. Finally, they observed that the effect of recombination can create particular cis-regulatory complexes that are able to mitigate deleterious recombination effects on regulatory circuits. Martin & Wagner [33] concluded that the effects of recombination may lead to many benefits, for example, increased genetic diversity and reduced genetic load, which are able to compensate for disadvantages caused by sexual reproduction.
Lohaus et al. [31] complemented results presented in Azevedo et al. [29] and MacCarthy & Bergman [32] by studying long-term benefits of sexual reproduction. Similarly, to previous studies [29,32], they observed sexual populations can evolve a higher robustness and a lower genetic load than asexual populations at equilibrium. However, contrary to Azevedo et al. [29], they found no evidence that negative epistasis can contribute to long- and short-term benefits emerged from sexual populations. Moreover, they found that a lower deleterious mutation rate evolves from sexual populations cannot sufficiently account for the ability of sexual populations to resist invasion by asexual populations in a long-term. Lohaus et al. [31] argued that it is the continuously increased recombinational robustness that minimises the cost of sexual reproduction, and ultimately evolves resistance to asexual invasion in a long term.
Wagner [34] broadly reviewed possible reasons of the low cost of recombination. He showed that
1. recombination can cause greater genotypic changes than mutation
2. recombination facilitates in creating new phenotypes
3. recombination is able to well preserve phenotypes in the context of genetic networks
4. recombination can preserve protein structure and function and
5. recombinational robustness could be substantially increased during evolution. Wagner therefore concluded that recombination can create new phenotypes while disrupting welladaptive phenotypes much less than mutation.
Le Cunff & Pakdaman [30] studied the relationship between individual-level evolutionary dynamics and population9 level survival probability in the face of genetic and demographic stochasticity. Here genetic stochasticity refers to fluctuations in genetic composition (variability) while demographic stochasticity refers to fluctuations in population size. Different from previous studies which employed the Wagner GRN model with a fixed evolution space, the population size is not fixed in each generation and the extinction could happen due to genetic and demographic stochasticity modelled in the system. Le Cunff & Pakdaman [30] found that recombination rate, initial population size and mutation rates can all influence population survival probability.
Whitlock et al. [36] investigated how Hill-Robertson interference affects the evolutionary origin and maintenance of sex in the context of populations with evolvable genetic architecture. Whitlock et al. [36] showed population size only contributes to the short-term advantage of sex. They further demonstrated that the deleterious mutation rate and recombination load are the two key properties of the genetic architecture that determine the long- and short-term advantages of sex.
Whitlock et al. [35] built on previous work in [36] and studied how costly sex could be maintained. Whitlock et al. [36] explicitly included two costs of recombination and migration loads in the simulation model and showed that population structure promotes the evolution of costly sex.
Plasticity and evolvability
Evolvability is the capacity of a population to produce heritable phenotypic variation to rapidly adjust to certain types of environmental challenges or opportunities [69-72]. This capacity, documented in nature, reflects phenotypic plasticity enabled by the capacity of evolution to capture and represent regularities not only in extant environments but in the ways in which the environments tend to change [15,73,74]. The simplest form of evolvability is simply variation - the rate of evolution is determined by the amount of variations in a population [75,76]. More sophisticated evolvability can be achieved via hierarchical complex organizations, for example, genetic networks [77-82]. Many previous studies have focused on reconciling the antagonistic relationship between robustness3 and evolvability by showing that living system can sustain phenotypic stability while producing genetic variations that lead to evolutionary innovations [24,69,83,84]. However, the concept of evolvability is still controversial, and how genetic networks evolve and become evolvable remains an open question [69,79].
FOOT NOTE
3Here robustness refers to the capacity to withstand mutations and maintain the phenotypic stability, function or structure
Bergman & Siegal [37] introduced gene `knock-out’ operation to Wagner’s GRN model and assessed phenotypic diversity before and after evolution. They showed that when a random gene is deleted by zeroing its corresponding row and column of the regulatory matrix in Wagner’s GRN model, environmental and genetic canalisation can both break down, but consequently the `knock-out’ operation increases the rate of adaptation to new environments. Moreover, they further conducted knock-out experiments on yeasts and found that they exhibit variations in phenotype which well matches their model predictions. Bergman and Siegal highlighted their results that complex genetic networks enable the evolutionary capacitor to buffer genotypic variations under normal conditions, while promoting the accumulation of hidden polymorphism that can facilitate new adaptations under stress.
Borenstein & Krakauer [38] looked at micro- and macroevolutionary patterns by evolving genotype-phenotype map in genetic networks. They showed that many evolutionary patterns observed and identified from empirical studies can be attributed to epistatic interactions between genes in regulatory networks. Borenstein and Krakauer [38] highlighted that their findings support the view that development is an essential component in the production of endless forms, and it is also critical to constrain biotic diversity and evolutionary trajectories.
Draghi & Wagner [42] studied whether natural selection facilitates the evolution of evolvability, particularly focusing on sexual populations. By introducing fluctuating environments (periodically changing optimal phenotypes), they demonstrated that natural selection facilitates the capacity of genetic networks to quickly adapt to new environments. This pattern was observed regardless of asexual or sexual reproduction modes, which suggests recombination does not suppress the evolution of evolvability. Draghi & Wagner [42] highlighted that the evolution of evolvability can be achieved by evolving complex genotype-phenotype map.
Fierst [43] investigated conditions under which a network may produce a more evolvable phenotype. Specifically, they modified Wagner’s GRN model by introducing a sexually dimorphic trait which has an underlying pleiotropic architecture that can affect evolvability. They showed that sexually dimorphic characters not only increase mutational robustness but also substantially facilitate evolvability. When she looked more closely to the results, Fierst [43] further found that linkage disequilibrium within or between sex is accounted for different levels of evolvability between sexually dimorphic and monomorphic populations.
Fierst [40] studied the effect of a history of phenotypic plasticity on adaptability to new environments. She found that populations with a history of phenotypic plasticity are able to adapt to new environments more rapidly than populations without a history of phenotypic plasticity, but the magnitude of the increased adaptation rate is dependent on the strength of selection in the original environments |Weak selection generally facilitates phenotypic plasticity, and substantially increases adaptation rate. Fierst [40] suggested that the results predict that the relative invasive capacity of different traits could be assessed through phenotypic variance in the original environment.
Espinosa-Soto et al. [39] introduced non-genetic perturbations (changes in gene initial expression patterns) and explored whether conditions under which phenotypic plasticity facilitate adaptation can be fulfilled in the context of genetic networks. They showed that non-genetic perturbations, such as gene expression noise, environmental changes, or epigenetic modifications can substantially stimulate phenotypic plasticity and ultimately facilitate adaptation to new environments. Espinosa-Soto et al. [39] concluded that the phenotypic plasticity has an essential role in adaptive evolution.
Pinho et al. [41] investigated how different levels of noise (changes in gene initial expression patterns as well as perturbations at the developmental stage) can affect the accessibility of phenotypic space that facilitates phenotypic diversity. They found that the increased levels of noise typically decrease accessibility to phenotypic space if the gene expression is binary but increase accessibility if there are more gene expression states. Pinho et al. [41] concluded that under specific conditions noise enables individuals to explore more phenotypic space.
Wilder & Stanley [44] compared the evolvability at the individual level with the evolvability at the population level, focusing on the potential of generating phenotypic variations. Specifically, by introducing the divergent selection, the selection for phenotypic variations, they showed that the divergent selection is able to produce evolvable populations and encourage phenotypic diversity, whereas evolvable individuals are more likely to be formed by the adaptive selection to fluctuating environments. Wilder & Stanley [44] hypothesized that non-adaptive mechanisms may be more important for shaping the emergence of evolvability.
Other Applications
The Wagner GRN model has also been employed to study the below research questions. Wagner [14] formally proposed a simple mathematical model to capture the key developmental process underlying transcriptional regulation and employed the proposed model to study the mechanism of gene duplication and its effect on phenotypic stability. He found that there is about 40%, at the highest, genes in a network are duplicated, depending on the fraction of genes that are duplicated in a single duplication event. Wagner [14] concluded that the evolution of gene networks should occur by gene duplications, and the most two favorable forms of genomic organization are tight linkage or strong dispersal.
Siegal et al. [45] first employed Wagner’s GRN model to thoroughly study the relationship between network topology and its functional of evolutionary properties. They found that the degree distribution (scale-free, power law distribution) itself does not have a major effect on functional properties associated with nodes. Moreover, there is a weak or nearly none correlation between network connectivity and genetic variations.
MacCarthy & Bergman [85] employed Wagner’s GRN model to study the sub functionalization indicated by the theory of duplication-degeneration-complementation. They showed that, in contrast to the previous theory predictions, sub functionalization and neofunctionalization can coexist in biological networks following gene duplication. MacCarthy & Bergman [85] hypothesized that this pattern is facilitated by the evolutionary plasticity in combination with the phenotypic neutrality which is prevailing in biological systems.
Sevim & Rikvold [51] studied the effect of the evolution of genetic robustness on the dynamical character of gene regulatory networks. Here the dynamical character refers to the phenotypic stability of genetic networks against perturbations, such as mutations or noise. They showed that stabilizing selection only weakly affects the network dynamical properties, and the networks that are most robust to mutations and noise are highly chaotic. Sevim & Rikvold [51] argued that the damage propagation analysis does not provide much useful information about robustness to mutations or noise in the context of genetic networks.
Palmer & Feldman [46] investigated the Bateson- Dobzhansky- Muller incompatibilities and extended Orr’s model [86,87] to account for the complex dynamics of incompatibility in the context of genetic networks. They showed that depending on certain model parameters, under constant selection environment, three patterns of system dynamics can be observed: hybrid incompatibility between two allopatric populations may,
a) Not increase at all
b) May increase to large values and
c) A pair of populations may `drift’ in and out of compatibility.
Espinosa-Soto & Wagner [48] investigated how modularity evolves in the context of genetic networks when the developmental process is explicitly modelled in the system. They showed that modularity is able to arise in genetic networks as a by-product of specialization in gene activity. They also demonstrated that new gene activity patterns that share existing patterns of gene activity are more likely favoured by the evolution of modularity.
Rhone et al. [49] studied the impact of selection on genes at the phenotypic level in the context of regulatory networks. They showed that there is a positive relationship between the selection strength on the phenotype and the level of regulation between the loci. Moreover, they found that genes that strongly regulate other genes as well as those are less regulated by other genes are responding more profoundly to selection within the network.
Pinho et al. [50] investigated how varying features and parameters of Wagner’s GRN model affect on network transition from oscillatory dynamics to developmental stability. They showed that the cyclic behaviour is mainly due to complex epistatic interactions between genes, but not due to connection strengths or patterns. Moreover, they showed that the stability distribution is highly robust to various model parameters and found that sparse networks are more likely to be stable.
Espinosa-Soto [47] studied how modularity evolves in gene regulatory networks. Espinosa-Soto [47] found that sparseness does not account for modularity in gene regulatory networks. However, the author argued that sparseness can enhance the 11 selection for multiple gene activity patterns.
Odorico et al. [52] investigated the influence of parental effects in the context of gene regulatory networks. Odorico et al. [52] demonstrated that adaptation and robustness can be enhanced for fast-developing individuals with when they were evolved under nongenetic inheritance.
Discussion & Future Work
The Wagner GRN model has been extensively used to explore many fundamental research questions in evolutionary biology and ecology. But only a few studies focus on the analysis of the system per se. In particular, due to the non-linear mapping from the regulatory response to the output phenotype at each time step during the self-developmental stage, it has been hard to determine whether the network is able to reach an equilibrium phenotypic state, or, if it could, what its equilibrium state would be. This is critical to many research questions. For example, the robustness assessed in most studies is to examine if the network remains stable when it is subject to certain perturbations. For research work on evolvability, researchers focus on studying whether the individual network can generate novel and inheritable phenotypes in the face of, for example, fluctuating environments. In most current studies, the equilibrium phenotypic state is examined or calculated through iterating the difference equations within certain time steps. This is, however, an extremely time-consuming solution, especially for evolving a large-size population for a very long evolution time. Since the self-developmental process can greatly slow down the simulation, it is worth exploring how to efficiently calculate or estimate the equilibrium phenotypic state analytically. In the meanwhile, the high-performance computing techniques need to be employed to further speed up the simulation process.
The currently available studies have implemented many different types of mutation or noise. For example, on the one hand, mutations happen in the genotype where they can change existing regulations by altering non-zero entries or change the network topology by creating new regulations in zero entries or deleting existing regulations from non-zero entries. The mutation can also occur in individual’s initial expression state and consequence alter its equilibrium state. The noise, on the other hand, is normally modelled at each time step during the self-development stage. However, to my best knowledge, the recombination operator has not yet been thoroughly explored. Most studies follow the `free recombination’ strategy. But we know that the offspring may not inherit an equal information from its parents, and there are many different mating strategies in nature. By implementing different recombination operators, we may be able to gain a better understanding of the origin and maintenance of sex and recombination for different species in nature. For example, by differentiating males and females in sexual lineages, we may be able to rigorously examine the role of sexual selection. We could also implement different features, such as different mutation rate, for males and females together with varying mating strategies to test if that would affect the underlying evolutionary dynamics.
Most of the current studies also have strictly required that each individual in the population is capable of achieving the developmental equilibrium. In other words, networks with oscillating phenotypic states will be wiped out immediately from the population. However, although this requirement is a reasonable biological assumption, it largely impedes alternative pathway evolution through, for example, compensatory mutations. In fact, many empirical studies have indicated that the fluctuating selection regime (periods of purifying selection) is also biologically realistic. In fact, networks that have re-gained the developmental stability may have very different properties compared with networks that have never been comprised. Those properties are expected to affect the underlying evolutionary dynamics. which have been largely overlooked in the current studies. Therefore, the future studies are encouraged to consider including those networks that have been through compensation in the simulation, and examine, if any, different evolutionary consequences.
However, if we would allow compromised networks to stay in the population for a while, then the problem would be, for example, how to calculate its fitness. The fitness evaluation functions used in current studies measure the phenotypic distance between the individual’s equilibrium state and a given target state. But if the individual is not able to achieve the developmental stability, then we cannot calculate individual fitness because there is no equilibrium state. In some studies, instead, the average expression state was used to calculate fitness for networks with oscillating phenotypic states. This is, however, a temporary expedient. In fact, the fitness evaluation should consider both individual’s ability to reach the developmental equilibrium and its distance away from the target. This is also biologically realistic as in many biological organizations. For example, in proteins, there is a balance between stability and function. Therefore, the future work is expected to take both network stability and its function into consideration when evaluating individual’s fitness.
The Wagner GRN model also has a great potential to be used to solve optimization problems in machine learning field since the model can converge to the target phenotype. It has been found that Darwinian process of mutation, recombination and selection are useful to study complex adaptations in evolutionary computation, a subfield of artificial intelligence [13,72,88]. Many computational evolutionary algorithms have been used to solve real-world engineering optimization problems [15,87,89]. For example, genetic algorithms (GAs) are methods well-suited for search and optimization in non-linear and high-dimensional problems Holland [90]. Convergence to near-optimal solutions is often perceived as the goal for GAs. Since the goal of Wagner’s GRN model is to find an optimal (target) phenotype, therefore it is possible to develop a similar system for discovering highly-evolvable genomes by exploiting genetic networks [91,92]. The many-to-one mapping mechanism of genotype to phenotype explicitly modelled in Wagner’s GRN model enables genes to buffer against and even exploit likely variations in the genome. In addition, such a dual learning system — coupled plasticity — is known to accelerate evolution in the right contexts [93,94].
Hinton & Nowlan [93] focused on the interaction between evolution and learning, showing that coupled plasticity can solve a problem that is extremely difficult for an evolutionary process on its own. Especially, the genotype used in Wagner’s GRN can be regarded as the hierarchical structures that control the network output (phenotype), i.e., represented as a possible solution to the problem [95]. Therefore, the aim is to explore how the robustness of genetic networks can improve the evolvability of evolutionary computation methods by exploiting genotypes for learning structure required for quick adaptations to environmental changes. Some preliminary results are presented in Wang et al. [15].
As George E. P. Box said, Essentially, all models are wrong, but some are useful.” The model developed under Wagner’s framework makes no attempt to fully cover biochemical processes of the underlying transcriptional regulation in real biological systems [96]. Instead, the abstraction of regulatory systems, as well as the self-developmental process, are explicitly modelled and emphasized [97]. The conclusions drawn from research using such an abstract model aim at providing useful high-level explanations or perditions for general patterns or properties that we would observe in natural systems [98].
References
- Karlebach G, Shamir R (2008) Modelling and analysis of gene regulatory networks. Nat Revi Mol Cell Biol 9(10): 770-780.
- Guelzim N, Bottani S, Bourgine P, Kepes F (2002) Topological and causal structure of the yeast transcriptional regulatory network. Nat Genet 31(1): 60-63.
- Lynch M (2007) The evolution of genetic networks by non-adaptive processes. Nature Reviews Genetics 8(10): 803-813
- Payne JL, Wagner A (2015) Mechanisms of mutational robustness in transcriptional regulation. Front in Genet 6: 322.
- Sorrells TR, Johnson AD (2015) Making sense of transcription networks. Cell 161(4): 714-723.
- Touch BB, Li H, Johnson AD (2008) Evolution of eukaryotic transcription circuits. Science 319(5871): 1797-1799.
- Wray GA, Hahn MW, Abouheif E, Balho JP, Pizer M, et al. (2003) The evolution of transcriptional regulation in eukaryotes. Molecular Biology and Evolution 20(9): 1377-1419.
- Ciliberti S, Martin OC, Wagner A (2007b) Robustness can evolve gradually in complex regulatory gene networks with varying topology. PLoS Comput Biol 3(2): e15.
- Fierst JL, Phillips PC (2015) Modeling the evolution of complex genetic systems: The gene network family tree. Journal of Experimental Zoology. Part B, Molecular and Developmental Evolution 324(1): 1-12.
- von Dassow G, Meir E, Munro EM, Odell GM (2000) The segment polarity network is a robust developmental module. Nature 406(6792): 188-192.
- Li F, Long T, Lu Y, Ouyang Q, Tang C (2004) The yeast cell-cycle network is robustly designed. Proc Natl Acad Scie USA 101(14): 4781-4786.
- Wagner A (1996) Does evolutionary plasticity evolve? Evolution 50(3): 1008-1023.
- Spirov A, Holloway D (2013) Using evolutionary computations to understand the design and evolution of gene and cell regulatory networks. Methods 62(1): 39-55.
- Wagner A (1994) Evolution of gene networks by gene duplications: A mathematical model and its implications on genome organization. Proceedings of the National Academy of Sciences of the United States of America 91(10): 4387-4391.
- Wang Y, Yin J (2014) Intelligent search optimized edge potential function (EPF) approach to synthetic aperture radar (SAR) scene matching. In Evolutionary Computation (CEC), IEEE Congress on, pp. 2124-2131.
- Wang Y (2015) Contourlet-based multispectral image fusion using free search differential evolution. In: Proceedings of the 12th Biennial International Conference on Artificial Evolution, Artificial Evolution Association, France, Europe, pp. 318-325.
- Ciliberti S, Martin OC, Wagner A (2007a) In-novation and robustness in complex regulatory gene net-works. Proceedings of the National Academy of Sciences of the United States of America 104(34): 13591-13596.
- Espinosa SC (2016) Selection for distinct gene expression properties favours the evolution of mutational robustness in gene regulatory networks. J Evol Biol 29(11): 2321-2333.
- Espinosa SC, Martin OC, Wagner A (2011b) Phenotypic robustness can increase phenotypic variability after nongenetic perturbations in gene regulatory circuits. J Evol Biol 24(6): 1284-1297.
- Huerta SE, Durrett R (2007) Wagner’s canalization model. Theor Popul Biol 71(2): 121-130.
- Kimbrell T, Holt RD (2007) Canalization breakdown and evolution in a source-sink system. Ame Nat 169(3): 370-382.
- Le Cun Y, Pakdaman K (2012) Phenotype genotype relation in Wagner’s canalization model. J Theo Biol 314: 69-83.
- Leclerc RD (2008) Survival of the sparsest: Robust gene networks are parsimonious. Mol Syst Biol 4: 213.
- Martin OC, Wagner A (2008) Multifunctionality and robustness tradeoffs in model genetic circuits. Biophys J 94(8): 2927-2937.
- Masel J (2004) Genetic assimilation can occur in the absence of selection for the assimilating phenotype, suggesting a role for the canalization heuristic. J Evol Biol 17(5): 1106-1110.
- Runneburger E, Le Rouzic A (2016) Why and how genetic canalization evolves in gene regulatory networks. BMC Evolutionary Biology 16(1): 239.
- Shin J, MacCarthy T (2015) Antagonistic coevolution drives Whack-AMole sensitivity in gene regulatory networks. PLoS Computational Biology 11(10): e1004432.
- Siegal ML, Bergman A (2002) Waddington’s canalization revisited: Developmental stability and evolution. Proce Natl Acad USA 99(16): 10528-10532.
- Azevedo RBR, Lohaus R, Srinivasan S, Dang KK, Burch CL (2006) Sexual reproduction selects for robustness and negative epistasis in artificial gene networks. Nature 440: 87-90.
- Le Cun Y, Pakdaman K (2014) Reproduction cost reduces demographic stochasticity and enhances inter-individual compatibility. Journal of Theoretical Biology 360: 263-270.
- Lohaus R, Burch CL, Azevedo RBR (2010) Genetic architecture and the evolution of sex. The Journal of Heredity 101(1): S142-S157.
- MacCarthy T, Bergman A (2007a) Coevolution of robustness, epistasis, and recombination favors asexual repro-duction. Proceedings of the National Academy of Sciences of the United States of America 104(31): 12801-12806.
- Martin OC, Wagner A (2009) Effects of recombination on complex regulatory circuits. Genetics 183(2): 673-684.
- Wagner A (2011) The low cost of recombination in creating novel phenotypes. Bio Essays 33(8): 636-646.
- Whitlock AOB, Azevedo RB, Burch CL (2018) Population structure promotes the evolution of costly sex in artificial gene networks. bioRxiv, USA.
- Whitlock AOB, Peck KM, Azevedo RBR, Burch CL (2016) An evolving genetic architecture interacts with hill-Robertson interference to determine the benefit of sex. Genetics 203(2): 923-936.
- Bergman A, Siegal ML (2003) Evolutionary capacitance as a general feature of complex gene networks. Nature 424(6948): 549-552.
- Borenstein E, Krakauer DC (2008) An end to endless forms: Epistasis, phenotype distribution bias, and nonuniform evolution. PLoS Comput Biol 4(10): e1000202.
- Espinosa SC, Martin OC, Wagner A (2011a) Phenotypic plasticity can facilitate adaptive evolution in gene regulatory circuits. BMC Evolutionary Biology 11(1): 5.
- Fierst JL (2011) A history of phenotypic plasticity accelerates adaptation to a new environment. Journal of Evolutionary Biology 24(9): 1992- 2001.
- Pinho R, Garcia V, Feldman MW (2015) Phenotype accessibility and noise in random threshold gene regulatory networks. PLoS ONE 10(4): e0119972.
- Draghi J, Wagner GP (2009) The evolutionary dynamics of evolvability in a gene network model. Journal of Evolutionary Biology 22(3): 599-611.
- Fierst JL (2010) Sexual dimorphism increases evolvability in a genetic regulatory network. Evolutionary Biology 38(1): 52-67.
- Wilder B, Stanley K (2015) Reconciling explanations for the evolution of evolvability. Adaptive Behavior 23(3): 171-179.
- Siegal ML, Promislow DEL, Bergman A (2007) Functional and evolutionary inference in gene networks: Does topology matter? Genetica 129(1): 83-103.
- Palmer ME, Feldman MW (2009) Dynamics of hybrid incompatibility in gene networks in a constant environment. Evolution 63(2): 418-431.
- Espinosa SC (2018) On the role of sparseness in the evolution of modularity in gene regulatory networks. PLOS Comput Biol 14(5): e10061172.
- Espinosa SC, Wagner A (2010) Specialization can drive the evolution of modularity. PLoS Comput Biol 6(3): e1000719.
- Rhone B, Brandenburg JT, Austerlitz F (2011) Impact of selection on genes involved in regulatory net-work: A modelling study. Journal of Evolutionary Biology 24(10): 2087-2098.
- Pinho R, Borenstein E, Feldman MW (2012) Most networks in Wagner’s model are cycling. PLoS One 7(4): e34285.
- Sevim V, Rikvold PA (2008) Chaotic gene regulatory networks can be robust against mutations and noise. Journal of Theoretical Biology 253(2): 323-332.
- Odorico A, Runneburger E, Le Rouzic A (2018) Modelling the influence of parental effects on gene-network evolution. Journal of Evolutionary Biology 31(5): 687-700.
- Waddington CH (1953) Genetic assimilation of an acquired character. Evolution 7(2): 118-126.
- Waddington CH (1959) Canalization of development and genetic assimilation of acquired characters. Nature 183(4676): 1654-1655.
- Bhattacharya S, Zhang Q, Andersen M (2011) A deterministic map of Waddington’s epigenetic landscape for cell fate specification. BMC Syst Biol 5: 85.
- Pujadas E, Feinberg AP (2012) Regulated noise in the epigenetic landscape of development and disease. Cell 148(6): 1123-1131.
- Gibson G, Wagner G (2000) Canalization in evolutionary genetics: A stabilizing theory? Bio Essays 22(4): 372-380.
- Eshel I, Feldman MW (1970) On the evolutionary effect of recombination. Theor popul Biol 1(1): 88-100.
- Feldman MW, Otto SP, Christiansen FB (1996) Population genetic perspectives on the evolution of recombination. Annu Revi Genet 30: 261- 295.
- Otto SP, Feldman MW (1997) Deleterious mutations, variable epistatic interactions, and the evolution of recombination. Theoretical Population Biology 51(2): 134-147.
- West, Lively, Read (1999) A pluralist approach to sex and recombination. Journal of Evolutionary Biology 12(6): 1003-1012.
- Hurst LD, Peck JR (1996) Recent advances in understanding of the evolution and maintenance of sex. Trends in Ecology & Evolution 11(2): 46- 52.
- Meirmans S, Strand R (2010) Why are there so many theories for sex, and what do we do with them? Journal of Heredity 101(1): S3-S12.
- Otto SP, Lenormand T (2002) Resolving the para-dox of sex and recombination. Nature Reviews Genetics 3(4): 252-261.
- Barton NH (2009) Why sex and recombination? Cold Spring Harbor Symposia on Quantitative Biology 74: 187-195.
- Kondrashov AS (1993) Classification of hypotheses on the advantage of amphimixis. Journal of Heredity 84(5): 372-387.
- Kouyos RD, Silander OK, Bonhoeer S (2007) Epistasis between deleterious mutations and the evolution of recombination. Trends Ecol Evol 22(6): 308-315.
- Otto SP, Gerstein AC (2006) Why have sex? The population genetics of sex and recombination. Biochemical Society Transactions 34(4): 519- 522.
- Masel J, Trotter MV (2010) Robustness and evolvability. Trends in Genetics 26(9): 406-414.
- Pigliucci M (2008) Is evolvability evolvable? Nature Reviews Genetics 9(1): 75-82.
- Wagner A (2007) Robustness and evolvability in living systems. Princeton studies in complexity. Princeton University Press, USA.
- Wagner GP, Altenberg L (1996) Perspective: Complex adaptations and the evolution of evolvability. Evolution 50(3): 967-976.
- Callahan HS, Pigliucci M, Schlichting CD (1997) Developmental phenotypic plasticity: Where ecology and evolution meet molecular biology. Bioessays 19(6): 519-525.
- Pigliucci M, Murren CJ, Schlichting CD (2006) Phenotypic plasticity and evolution by genetic assimilation. J Exp Biol 209(12): 2362-2367.
- Fisher RA (1930) The Genetical Theory of Natural Selection. Oxford University Press, UK.
- Price GR (1972) Fisher’s `fundamental theorem’ made clear. Ann Hum Genet 36(2): 129-140.
- Aldana M, Balleza E, Kauffman S, Resendiz O (2007) Robustness and evolvability in genetic regulatory networks. J Theor Biol 245(3): 433- 448.
- Clune J, Mouret JB, Lipson H (2013) The evolutionary origins of modularity. Proceedings of the Royal Society of London B: Biological Sciences 280: 1755.
- Crombach A, Hogeweg P (2008) Evolution of evolvability in gene regulatory networks. PLoS Computational Biology 4(7): e1000112.
- Greenbury SF, Johnston IG, Smith MA, Doye JPK, Louis AA (2010) The effect of scale-free topology on the robustness and evolvability of genetic regulatory net-works. J Theor Biol 267(1): 48-61.
- Landry CR, Lemos B, Rifkin SA, Dickinson WJ, Hartl DL (2007) Genetic properties influencing the evolvability of gene expression. Science 317(5834): 118-121.
- Torres SC, Huang S, Aldana M (2012) Criticality is an emergent property of genetic networks that exhibit evolvability. PLoS Computational Biology 8(9): e1002669.
- Garefield DA, Runcie DE, Babbitt CC, Haygood R, Nielsen WJ, et al. (2013) The impact of gene expression variation on the robustness and evolvability of a developmental gene regulatory network. PLoS Biology 11(10): e1001696.
- Whitacre J, Bender A (2010) Degeneracy: A design principle for achieving robustness and evolvability. Journal of Theoretical Biology 263(1): 143-153.
- MacCarthy T, Bergman A (2007b) The limits of sub functionalization. BMC Evolutionary Biology 7(1): 213.
- Orr HA (1995) The population genetics of speciation: The evolution of hybrid incompatibilities. Genetics 139(4): 1805-1813.
- Orr HA (1996) Dobzhansky, bateson and the genetics of speciation. Genetics 144(4): 1331-1335.
- Yin J, Wang Y, Hu J (2012) Free search with adaptive differential evolution exploitation and quantum-inspired exploration. Journal of Network and Computer Applications 35(3): 1035-1051.
- Yin J, Wang Y, Hu J (2012b) A new dimensionality reduction algorithm for hyperspectral image using evolutionary strategy. Industrial Informatics. IEEE Transactions on 8(4): 935-943.
- Holland JH (1992) Adaptation in natural and artificial systems: an introductory analysis with applications to biology, control and artificial intelligence. MIT Press, Cambridge, MA, USA.
- Payne JL, Moore JH, Wagner A (2014) Robustness, evolvability, and the logic of genetic regulation. Artificial Life 20(1): 111-126.
- van Dijk ADJ, van Mourik S, van Ham RCH J (2012) Mutational robustness of gene regulatory networks. PLoS ONE 7(1): e30591.
- Hinton GE, Nowlan SJ (1987) How learning can guide evolution. Complex Systems 1(3): 495-502.
- Kashtan N, Noor E, Alon U (2007) Varying environments can speed up evolution. Proc Natl Acad Sci USA 104(34): 13711-13716.
- Felix MA, Barkoulas M (2015) Pervasive robustness in biological systems. Nat Revi Genet 16(8): 483-496.
- Wagner A (2008) Robustness and evolvability: A paradox resolved. Proceedings of the Royal Society of London B: Biological Sciences 275(1630): 91-100.
- Wang Y, Lan Y, Weinreich DM, Priest NK, Bryson JJ (2015) Recombination is surprisingly constructive for artificial gene regulatory networks in the context of selection for developmental stability. In: Proceedings of the European Conference on Artificial Life 2015, The MIT Press, USA, pp. 530-537.
- Wang Y, Matthews SG, Bryson JJ (2014) Evolving evolvability in the context of environmental change: A gene regulatory network (GRN) approach. In Artificial Life 14: International Conference on the Synthesis and Simulation of Living Systems. The MIT Press, USA, pp. 47-53.
© 2019 Yifei Wang. This is an open access article distributed under the terms of the Creative Commons Attribution License , which permits unrestricted use, distribution, and build upon your work non-commercially.
Editor In Chief







.jpg)

























Signup for Newsletter

Quick Links
 Editorial Board Registrations
Editorial Board Registrations Submit your Article
Submit your Article Refer a Friend
Refer a Friend Advertise With Us
Advertise With UsOur Recent Edition

.jpg)



Top Editors




.jpg)

















.bmp)
.jpg)
.png)
.jpg)













.jpg)






.png)

.png)




.png)




Financial Support

Sponsors

Latest e-Books

Latest Video

 a Creative Commons Attribution 4.0 International License. Based on a work at www.crimsonpublishers.com.
Best viewed in
a Creative Commons Attribution 4.0 International License. Based on a work at www.crimsonpublishers.com.
Best viewed in