
- About Us
- Information
-

The Author ensures that the research has been conducted responsibly and ethically with adherence to all relevant regulations. read more..
- For Authors
- For Reviewer
- Manuscript Guidelines
- Membership
- Publication Ethics
-
- Journals
- Reprints
- e-Books
- Videos
- Policies
- Contact Us
 COVID-19
COVID-19
- Submissions



Full Text
Cohesive Journal of Microbiology & Infectious Disease
Computational Prediction for Antibiotics Resistance Through Machine Learning and Pk/Pd Analysis
Hyunjo Kim1* and Jaehoon Song2
1School of pharmacy, Yonsei university, South Korea
2ANSORP, CHA Bio Group at CHA Bio complex, South Korea
*Corresponding author:Hyunjo Kim, School of pharmacy, Yonsei university, Campus town, Songdo techno park, Incheon city, South Korea
Submission: June 13, 2019 ; Published: June 24, 2019

ISSN 2578-0190 Volume2 Issue5
Abstract
Infection disease is a major cause of morbidity and mortality in the developing world. Antibiotics resistance, which is predicted to rise in many countries worldwide, threatens infection treatment and control. The machine learning can help to identify patients at higher risk of treatment failure in infection diseases Closer monitoring of these patients may decrease treatment failure rates and prevent emergence outbreak of antibiotic resistance. To identify features associated with treatment failure and to predict which patients are at highest risk of antibiotics resistance this study was designed. The most predictive model was forward stepwise selection, although most models performed at or above targeted value. We employed a range of powerful machine learning tools to predict antibiotic resistance from whole genome sequencing data for model strain. We used the presence or absence of genes, population structure and isolation year of isolates as predictors, and could attain average precision and recall without prior knowledge about the causal mechanisms. These results demonstrate the potential application of machine learning methods as a diagnostic tool in healthcare settings.
Keywords: Infection disease; Antibiotics resistance; Machine learning model algorithm; Genome analysis; Antibiotics resistance genes (ARGs); PK/PD analysis
Introduction
An alarming and persistent rise in antibiotic resistance among many important pathogenic bacterial species poses one of the greatest contemporary challenges to the public health. The treatment of bacterial infections is becoming increasingly ineffective due to rapid mutation which leads to antibiotic resistant and resistant bacteria become more prevalent. As result the existing antibiotics are gradually obsolete and again new drugs are needed to be designed for the same threat. Recently, antibiotic resistance is a global health crisis linked to increased, and often unrestricted, antibiotic use in humans and animals [1]. The rise of multiclass-antibiotics resistant pathogens and the dearth of new antibiotic development place an existential strain on successful infectious disease therapy. Breakthrough strategies that go beyond classical antibiotic mechanisms are needed to combat this looming public health catastrophe. Reconceptualizing antibiotic therapy in the richer context of the host-pathogen interaction is required for innovative solutions. Here we review these relationships and their relevance to antimicrobial resistance (AMR) trends witnessed in the clinical setting. This review highlights the issues of enrichment and dissemination of antibiotics resistance genes (ARGs) [2,3] in the environment, and also future needs in mitigating the spread of antibiotic resistance in the environment, particularly under the planetary health perspective, i.e., the systems that sustain or threaten human health. By defining specific virulence factors, the essence of a pathogen, and pharmacologically neutralizing their activities, one can block disease progression and sensitize microbes to immune clearance. Likewise, host-directed strategies to boost phagocyte bactericidal activity, enhance leukocyte recruitment, or reverse pathogeninduced immunosuppression seek to replicate the success of cancer immunotherapy in the field of infectious diseases. The answer to the threat of multidrug-resistant pathogens lies in current antibiotic paradigms. Furthermore, the rise of bacterial strains resistant to multiple antibiotics is expected to dramatically limit treatment effectiveness, leading to potentially incurable outbreaks [4]. In addition to new drug development efforts, there is an urgent need for preclinical tools that are capable of effective and rapid detection of resistance, as culturebased laboratory diagnostics test are usually time consuming and costly. One of the major health threats of 2st century is emergence of antibiotic resistance. To manage its human health and economic impact, efforts are made to develop novel diagnostic tools that rapidly detect resistant strains in clinical settings [5,6]. In our study, we employed a range of powerful machine learning tools to predict antibiotic resistance from whole genome sequencing data for model bacteria. We used the designed machine learning algorithms as predictors, and could train average precision of the aimed value and recall of the closest number to mean without prior knowledge about the causal mechanisms considering environmental factors [7-9] related to key parameters such as antibiotic resistomes [10-14] and their mechanisms [15,16]. These results demonstrate the potential application of machine learning methods as a diagnostic tool in healthcare settings. Therefore, systemically designed algorithms based strategies to mitigate the spread of antibiotic resistance are suggested [17,18].
Overall, this article provides a conceptual framework for understanding the complexity of the problem of emergence of antibiotic resistance in the clinic. Availability of such knowledge will allow researchers to build models for dissemination of resistance genes and for developing interventions to prevent recruitment of additional or novel genes into pathogens.
Investigational Background
The successful treatment of infectious diseases heavily relies on the therapeutic usage of antibiotics. However, the high use of antibiotics in humans and animals leads to increasing pressure on bacterial populations in favorite of resistant phenotypes. Antibiotics reach the environment from a variety of emission sources and are being detected at relatively low concentrations. Given the possibility of selective pressure to occur at sub-inhibitory concentrations, the ecological impact of environmental antibiotic levels on microbial communities and resistance levels is vastly unknown. Quantification of antibiotics resistance genes (ARGs) and of antibiotic concentrations is becoming commonplace. Yet, these two parameters are often assessed separately and in a specific spatiotemporal context, thus missing the opportunity to investigate how antibiotics and ARGs relate. Furthermore, antibiotics (multi) resistance has been receiving ever growing attention from researchers, policy-makers, businesses and civil society. Our aim was to collect the limited data on antibiotic concentrations and ARGs abundance currently available to explore if a relationship could be defined. A metric of antibiotic selective pressure, i.e. the sum of concentrations corrected for microbial inhibition potency, was used to correlate the presence of antibiotics in the environment to total relative abundance of ARGs while controlling for basic sources of non-independent variability, such as strain and antibiotic class. The results of this meta-analysis show a significant statistical effect of antibiotic pressure and type of environmental compartment on the increase of ARGs abundance even at very low levels. Moreover, our analysis emphasizes the importance of integrating existing information particularly when attempting to describe complex relationships with limited mechanistic understanding in our previous paper (Figure 1).
Figure 1:Schematic illustration of antibiotics resistance investigation.

Methods
We conduct a systematic review, selecting studies if they are published randomized controlled trials (RCTs) which report the relationship between taking any antibiotics for any indication and incidence of resistant Strains in patients of any age group. We use a predefined search strategy to identify studies meeting these eligibility criteria in MEDLINE, Embase, Global Health and the Cochrane Central Register of RCTs. Two authors independently screen titles and abstracts, review the full texts and undertake data extraction. If feasible, we will perform pair-wise meta-analysis modelling to determine the relationship between the duration of antibiotics treatment and development of resistant Strain. If the identified studies meet the assumptions for a network metaanalysis, we additionally model this relationship using indirect comparisons. This hypothesis illustrated in Figure 2 as an example for macrolide class and its hypothetical network of anticipated randomized controlled trial data for the effect of macrolide treatment duration on the development of antimicrobial resistance is presented. As shown in Figure 2, each treatment group is a node. The lines joining nodes, termed edges, will be drawn to thickness that graphically represents the amount of direct evidence: the number of comparisons that we expect to find between a particular pair of nodes.
Figure 2:Hypothetical network of anticipated randomised controlled trial data for the effect of chosen antibiotics treatment duration on the development of antimicrobial resistance.

Mechanisms of antibiotic resistance: genetic basis
Emergence of resistance among the most important bacterial pathogens is recognized as a major public health threat affecting humans worldwide [19-22]. Multidrug-resistant organisms have not only emerged in the hospital environment but are now often identified in community settings, suggesting that reservoirs of antibiotics resistant bacteria are present outside the hospital [23- 25]. The bacterial response to the antibiotic attack is the prime example of bacterial adaptation and the pinnacle of evolution. Survival of the fittest is a consequence of an immense genetic plasticity of bacterial pathogens that trigger specific responses that result in mutational adaptations, acquisition of genetic material, or alteration of gene expression producing resistance to virtually all antibiotics currently available in clinical practice [26-28]. Therefore, understanding the biochemical and genetic basis of resistance is of paramount importance to design strategies to curtail the emergence and spread of resistance and to devise innovative therapeutic approaches against multi-antibiotics resistant organisms.
Overview on evolutionary processes of the antibiotic resistance
Figure 3:Overview evolutionary processes of the antibiotic resistance.

During millions of years antibiotics and antibiotic resistance genes have co-evolved slowly. In this long period the first transition was the acquisition of pre-resistance genes by different bacteria. This genetic transference allowed the evolution toward true and more efficient antibiotic resistance genes. However, the great evolutionary transition was the discovery, mass production and consumption of antibiotics. Antibiotics accelerated dramatically the diversification of resistance genes and selection for reaching extraordinary efficient variants (Figure 3). In this study, we will describe in detail the major mechanisms of antibiotic resistance encountered in clinical practice, providing specific examples in relevant bacterial pathogens (Table 1).
Table 1:Different examples of resistance mechanisms with clinical relevance from natural functions in environmental bacteria.

Nowadays, the high-throughput sequencing tools and bioinformatics software, knowledge on high bacterial diversity in bacterial communities (metagenome) is increasing. A huge diversity of resistance mechanisms to practically all antibiotic families has been found in both antibiotic- and non-antibiotic-producing bacteria. Three types of resistomes can be defined: intrinsic, environmental, and unknown [29-33]. In the intrinsic resistome or pre-resistome, the antibiotic resistant elements belong to bacterial metabolic networks, reflecting their role in microbial physiology. They might be coupled to signaling molecules (antibiotics) facilitating the co-selection of antibiotics and antibiotic resistance genes in a constant arms-race over a long time. The intrinsic resistome is a wider concept and probably universal to the bacterial world. The description of the intrinsic resistome has expanded our knowledge about potential new resistance mechanisms [34-36]. They could become true antibiotic resistance genes if appropriate driving forces were exerted. Moreover, the potential adaptiveness of these pre-resistome genes can be accelerated if, by chance, they are transferred to new genetic contexts (Figure 3), where these genes may evolve toward more efficient enzymes without having a physiological role. As a consequence, this silent and non-predictive resistome (unknown resistome) is ready to be selected [37-39].
Mutational resistance gene transfer
Figure 4:Schematic representation of the mechanism of action and resistance to linezolid. Panel A. Linezolid interferes with the positioning of aminoacyl-tRNA; Panel B. Representation of domain V of 23S rRNA.

Bacteria have a remarkable genetic plasticity that allows them to respond to a wide array of environmental threats, including the presence of antibiotic molecules that may jeopardize their existence [40,41]. As mentioned, bacteria sharing the same ecological niche with antimicrobial-producing organisms have evolved ancient mechanisms to withstand the effect of the harmful antibiotic molecule and, consequently, their intrinsic resistance permits them to thrive in its presence [42,43]. From an evolutionary perspective, bacteria use two major genetic strategies to adapt to the antibiotic attack, mutations in gene(s) often associated with the mechanism of action of the compound, and acquisition of foreign DNA coding for resistance determinants through horizontal gene transfer [44,45].
In this scenario, a subset of bacterial cells derived from a susceptible population develop mutations in genes that affect the activity of the drug, resulting in preserved cell survival in the presence of the antimicrobial molecule. Once a resistant mutant emerges, the antibiotic eliminates the susceptible population and the resistant bacteria predominate. In many instances, mutational changes leading to resistance are costly to cell homeostasis and are only maintained if needed in the presence of the antibiotic. In general, mutations resulting in antimicrobial resistance alter the antibiotic action via one of the following mechanisms (Figure 4).
Mutation of target sites
One of the most efficient mechanisms for accumulating antimicrobial resistance genes is represented by integrons, which are site-specific recombination systems capable of recruiting open reading frames in the form of mobile gene cassettes. Integrons provide an efficient and rather simple mechanism for the addition of new genes into bacterial chromosomes, along with the necessary machinery to ensure their expression; a robust strategy of genetic interchange and one of the main drivers of bacterial evolution. Thus, resistance arising due to acquired mutational changes is diverse and varies in complexity [46-50]. Classically, bacteria acquire external genetic material through three main strategies, transformation (incorporation of naked DNA), transduction (phage mediated) and, conjugation. Emergence of resistance in the hospital environment often involves conjugation, a very efficient method of gene transfer that involves cell-to-cell contact and is likely to occur at high rates in the gastrointestinal tract of humans under antibiotic treatment. As a general rule, conjugation uses mobile genetic elements (MGEs) as vehicles to share valuable genetic information, although direct transfer from chromosome to chromosome has also been well characterized. The most important MGEs are plasmids and transposons, both of which play a crucial role in the development and dissemination of antimicrobial resistance among clinically relevant organisms. In other words, antibiotics resistance mechanisms are summarized as following; modifications of the antimicrobial target (decreasing the affinity for the antibiotics), a decrease in the drug uptake, activation of efflux mechanisms to extrude the harmful molecule, or global changes in important metabolic pathways via modulation of regulatory networks [51].
For an instance, the ErmC leader peptide (Figure 5) is produced and the ermC mRNA forms two hairpins, preventing the ribosome to recognize the ribosomal binding site (RBS) of ermC. As a result, translation is inhibited under non-induction conditions. After exposure to erythromycin, the antibiotic interacts with the ribosome and binds tightly to the leader peptide, stalling progression of translation. This phenomenon releases the ermC RBS and permits translation.
Figure 5:Schematic representation of the post-transcriptional control of the gene. RBSL, ribosomal(blue) binding site of the leader; RBSC, ribosomal binding site of ermC; AUG, initiation codon.

Therefore, bacteria have evolved a sophisticated mRNA-based control mechanism to tightly regulate the expression of these methylases, ensuring a high efficiency of action in the presence of the antibiotic while minimizing the fitness costs for the bacterial population. Similarly, the system is usually not induced by lincosamides [46,47] or streptogramins [48]. However, the use of these agents against isolates carrying inducible erm genes may result in the selection of constitutive mutants in vivo (particularly in severe infections), leading to therapeutic failures.
Prediction of antibiotic resistance
The emergence of microbial antibiotic resistance is a global health threat. In clinical settings, the key to controlling spread of resistant strains is accurate and rapid detection. As traditional culture-based methods are time consuming, genetic approaches have recently been developed for this task. The detection of antibiotics resistance is typically made by measuring a few known determinants previously identified from genome sequencing, and thus requires the prior knowledge of its biological mechanisms. To overcome this limitation, we employed machine learning models to predict resistance to chosen compounds across multi-classes of antibiotics from existing and novel whole genome sequences of model strains [17]. It was considered a range of methods, and examined population structure, isolation year, gene content, and polymorphism information as predictors. Gradient boosted decision trees consistently outperformed alternative models with an average accuracy of the aimed value on held-out data. While the best models most frequently employed gene content, an average accuracy scores could be obtained using population structure information alone. Single nucleotide variation data were less useful, and significantly improved prediction only for chosen antibiotics and its combination through given model algorithms through modification specially. These results demonstrate that antibiotics resistance can be accurately predicted from whole genome sequences without a strain knowledge of mechanisms, and that both genomic and epidemiological data can be informative. This paves way to integrating machine learning approaches into diagnostic tools in the clinic [18]. Still, the understanding of mode of evolution of resistance in bacteria is a determining step in the preclinical development of new antibiotics, because drug developers assess the risk of resistance arising against a drug during preclinical development.
Antibiotics resistance measurements index
The aim of the study was to evaluate a cumulative antibiotics resistance index (ARI) as a possible key outcome measure of antimicrobial stewardship programs and as a tool to predict antimicrobial resistance trend. Antibiotic susceptibility for model strains pathogens, recovered from blood cultures during a fixed time period, was analyzed to obtain a cumulative ARI. For each antibiotic tested a score of 0, 0.25, 0.5, 0.75 or 1 was assigned for susceptibility, intermediate resistance, or resistance, respectively, and the ARI was calculated by dividing the sum of these scores by the number of antibiotics tested. Cumulative ARI of microorganisms were compared, and a mathematical prediction model for antimicrobial resistance trend was obtained. ARI could be a useful tool to measure the impact of antibiotics surveillance programs on antibiotics resistance. This clinical prediction rule performed well with internal validation. It showed good calibration and good discrimination. The receiver operating curve is shown in Figure 6.
Figure 6:Schematic representation of the post-transcriptional control of the gene. RBSL, ribosomal(blue) binding site of the leader; RBSC, ribosomal binding site of ermC; AUG, initiation codon.

Predicting antibiotic resistance from resistance genes.
It has been developed a high-throughput multiplex PCR test for several resistance genes encoding OpGen. OpGen offers the high-throughput multiplex PCR test as a commercial testing service for analysis of bacterial isolates. The resistance genes were chosen based on a review of the scientific literature and surveys of resistance gene databases. PCR assays specific for gene variants at the amino acid positions indicated or homologous sequence regions shared by different gene subtypes within a family of closely related resistance genes were designed in Table 2. The resistance genes in database effectively covered the resistance mechanisms for the isolates in this study and could be used for the sensitive prediction of phenotypic resistance with generalized linear models, which allowed the inference of phenotypic susceptibility in the absence of predicted resistance. The binary models effectively predicted phenotypic resistance based on the presence of resistance genes but did not have sufficient sensitivity to infer phenotypic susceptibility in the absence of resistance genes because the binary models lacked full coverage of resistance mechanisms.
Table 2:Percentage of isolates with non-susceptible phenotypes per antibiotic.

Additionally, genome retrieval and identification analyses were performed. The complete genome of Strain sequences was downloaded from The National Center for Biotechnology Information (NCBI) Genome Database (https://www.ncbi.nlm. nih.gov/genome). The fasta file format of the genome sequence of 11 strains of bacteria were thoroughly analysed for Antibiotics Resistance Genes (ARGs) on the bulk analysis Resistance Gene Identifier (RGI) or CARD 2017 Platform (https://card.mcmaster. ca/ analyze/rgi). Default select criteria, which identified gene base on strict or perfect only was used. On the RGI platform each genome sequence file was uploaded and all settings were left at default. To have an inter-relation as well as qualitative and quantitative pattern of these ARGs in the various strains, a heatmap chart was constructed using Microsoft Excel 2016 version for Mac.
Mathematical modeling
All sequencing reads sets were used as inputs to each of antibiotics resistance prediction algorithms. In case of no calls or if there were discordant results among the reads sets of a sample for antibiotics, the prediction for the sample for that particular antibiotics was treated as missing. In our sensitivity and specificity calculation and parameter estimation for DST [52,53] credibility computation, which are subsequently described in detail, we omitted those samples clearly indicated to have been used to train the tools. The sensitivities and specificities of the algorithms for antibiotics were first calculated together with their 95% confidence interval, with the corresponding phenotypes in the data collection as the gold standard. Bioinformatics tools have also been developed to predict drug resistance from whole genome sequencing (WGS) data. Sufficiently large databases possessing both WGS and DST information have allowed the drug resistance to several antibiotics to be accurately predicted, to the extent that rapid diagnostics that target these mutations have been developed [54-57].
The DST credibility score for each sample was calculated as the probability specifically, for antibiotics i, let the prevalence of antibiotics resistance be Pi, and the true sensitivity, the true specificity, the proportion of no-call predictions among truly resistant samples and the proportion of no-call predictions among truly susceptible samples of the program be Sensij, Specij, NoRij, and NoSij, for j in {StrainProfiler, Anibiotics}. We denote the predictions of the algorithms as
𝑝𝑟𝑒𝑑𝑖𝑐𝑡𝑖𝑜𝑛𝑠 =𝑌𝑗𝑗 ∈ {Strain𝑃𝑟𝑜𝑓𝑖𝑙𝑒𝑟, Antibiotics},
where Yj = 1 if the sample is predicted resistant, 0 if susceptible,
and NA otherwise. The probability of the sample being resistant
given the predictions of the four tools is as follows:
𝑃 (𝑠𝑎𝑚𝑝𝑙𝑒 𝑖𝑠 𝑟𝑒𝑠𝑖𝑠𝑡𝑎𝑛𝑡 | 𝑝𝑟𝑒𝑑𝑖𝑐𝑡𝑖𝑜𝑛𝑠) = 𝑃 (𝑝𝑟𝑒𝑑𝑖𝑐𝑡𝑖𝑜𝑛𝑠 |
𝑠𝑎𝑚𝑝𝑙𝑒 𝑖𝑠 𝑟𝑒𝑠𝑖𝑠𝑡𝑎𝑛𝑡) / 𝑃𝑖𝐿𝑖𝑘𝑒𝑙𝑖ℎ𝑜𝑜𝑑 (𝑝𝑟𝑒𝑑𝑖𝑐𝑡𝑖𝑜𝑛𝑠),
with Likelihood(predictions) calculated as
𝑃 (𝑝𝑟𝑒𝑑𝑖𝑐𝑡𝑖𝑜𝑛𝑠 | 𝑠𝑎𝑚𝑝𝑙𝑒 𝑖𝑠 𝑟𝑒𝑠𝑖𝑠𝑡𝑎𝑛𝑡).
𝑃𝑖 + 𝑃 (𝑝𝑟𝑒𝑑𝑖𝑐𝑡𝑖𝑜𝑛𝑠 | 𝑠𝑎𝑚𝑝𝑙𝑒 𝑖𝑠 𝑠𝑢𝑠𝑐𝑒𝑝𝑡𝑖𝑏𝑙𝑒).
(1−𝑃𝑖) = 𝑗𝑃 (𝑌𝑗 | 𝑠𝑎𝑚𝑝𝑙𝑒 𝑖𝑠 𝑟𝑒𝑠𝑖𝑠𝑡𝑎𝑛𝑡).
𝑃𝑖 + 𝑗𝑃 (𝑌𝑗 | 𝑠𝑎𝑚𝑝𝑙𝑒 𝑖𝑠 𝑠𝑢𝑠𝑐𝑒𝑝𝑡𝑖𝑏𝑙𝑒). (1−𝑃𝑖),
where
𝑃 (𝑌𝑗 | 𝑠𝑎𝑚𝑝𝑙𝑒 𝑖𝑠 𝑟𝑒𝑠𝑖𝑠𝑡𝑎𝑛𝑡) = {𝑆𝑒𝑛𝑠𝑖𝑗 𝑖𝑓 𝑌𝑗 = 1,
1−𝑆𝑒𝑛𝑠𝑖𝑗−𝑁𝑜𝑅𝑖𝑗 𝑖𝑓 𝑌𝑗=0,
𝑁𝑜𝑅𝑖𝑗 𝑖𝑓 𝑌𝑗 𝑖𝑠 𝑁𝐴,
and
𝑃 (𝑌𝑗 ∣ 𝑠𝑎𝑚𝑝𝑙𝑒 𝑖𝑠 𝑠𝑢𝑠𝑐𝑒𝑝𝑡𝑖𝑏𝑙𝑒) = {1−𝑆𝑝𝑒𝑐𝑖𝑗−𝑁𝑜𝑆𝑖𝑗 𝑖𝑓 𝑌𝑗=1
𝑆𝑝𝑒𝑐𝑖𝑗 𝑖𝑓 𝑌𝑗=0
𝑁𝑜𝑆𝑖𝑗 𝑖𝑓 𝑌𝑗 𝑖𝑠 𝑁𝐴.
The DST credibility score of a sample thus equals P (sample is resistant | predictions) if its reported phenotype is resistant, or 1 – P (sample is resistant | predictions) if the reported phenotype is susceptible.
Prediction on antibiotic resistance at the PK/PD point of view
Antibiotic resistance constitutes one of the most pressing public health concerns [58-61]. Antimicrobial peptides (AMPs) of multicellular organisms are considered part of a solution to this problem, and AMPs produced by bacteria such as colistin are lastresort drugs. Importantly, AMPs differ from many antibiotics in their pharmacodynamic characteristics. Here we implement these differences within a theoretical framework to predict the evolution of resistance against AMPs and compare it to antibiotic resistance. Our analysis of resistance evolution finds that pharmacodynamic differences all combine to produce a much lower probability that resistance will evolve against AMPs. The finding can be generalized to all drugs with pharmacodynamics similar to AMPs [62-65]. Pharmacodynamic concepts are familiar to most practitioners of medical microbiology, and data can be easily obtained for any antibiotics or antibiotics combination. Our theoretical and conceptual framework is, therefore, widely applicable and can help avoid resistance evolution if implemented in antibiotic stewardship schemes or the rational choice of new antibiotics candidates [66].
Mutant selection window and pharmacodynamic parameters [67,68]
Schematically, the revised mutant selection window (MSW) and pharmacodynamic parameters are shown in Figure 7. The MSW is defined as the antimicrobial concentration range in which resistant mutants are selected [69,70-71]. Following MSW using net growth curves of a susceptible strain S and a resistant strain R are also determined. Mathematically, net growth is described with the pharmacodynamic function ψ(a). In short, the function consists of the four pharamcodynamic parameters [67,68,72-74]: net growth in the absence of antibicrobials ψ ax, net growth in the presence of a dose of antimicrobials, which effects the growth maximal, ψmin, the minimum inhibitory concentration (MIC) and the parameter κ, which describes the steepness of the pharamcodynamic curve. Here, the two pharmacodynamics functions ψS(a) and ψR(a) describe the net growth of the S and R, respectively, in relation to the drug concentration a. Cost of resistance c is included as a reduction of the maximum growth rate of the resistant strain ψmax,R, with c = 1-ψmax, R/ψmax,S. Note that with this definition, cost of resistance is expressed as reduction in net growth rate in the absence of antimicrobials (a = 0). The lower bound of the MSW is the concentration for which the net growth rate of the resistant strain is equal to the net growth rate of the sensitive strain and is called the minimal selective concentration (MSC). The upper bound is given by the MIC of the resistant strain MICR. It is calculated the size of the MSW as: . Following the original approach to define the MSW, the boundaries of the MSW can also be applied to the pharmacokinetics of the system. The width of the MSW is partly determined by the steepness of the pharmacodynamic curve (Figure 7).
Figure 7:Schematic representation of the post-transcriptional control of the gene. RBSL, ribosomal(blue) binding site of the leader; RBSC, ribosomal binding site of ermC; AUG, initiation codon.

Mathematical modeling focused on Pharmacodynamics
To study resistance evolution, we used a mathematical model [75,76] that incorporates pharmacokinetics (PK)/ pharmacodynamics (PD) [77] and captures population dynamics of bacterial populations under treatment with antimicrobial drugs. We ran stochastic simulations to calculate the probability of resistance emergence, the probability of takeover by a resistant strain, the time to resistance emergence and the risk of resistance. Importantly, the concentration range between no killing and maximal killing is much narrower for AMPs than antibiotics, resulting in a much steeper curve. The maximum killing rate of AMPs is much higher than of antibiotics, as reflected in quicker killing time. These differences between AMPs and antibiotics with respect to their pharmacodynamic parameters determine the size of the MSW and enable us to assess the influence of the MSW on resistance evolution. Another difference relevant to the evolution of resistance is the finding that many antibiotics increase mutation rates of bacteria, but the AMPs tested so far do not show such an effect as they do not elicit bacteria DNA repair responses [78,79].
Here, we use a pharmacodynamics approach that has been widely used to describe sigmoid dose-response relationships to study the evolution of resistance in a homogeneous population. Our work uses the formulation of pharmacodynamic function. We particularly explored how the steepness of the pharmacodynamic curve, together with other pharmacodynamic parameters determine the probability of resistance emergence [80,81].
The size of the MSW depends on the lower and upper bound of the MSW and is calculated as ratio, due to the logarithmic scale that is used to plot dose–response relationships;
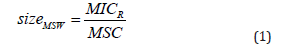
To analytically describe the MSW, we use the pharmacodynamic function ψ(a), which mathematically describes the net growth rate with a Hill function:
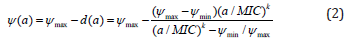
The analytic solution of the MSC is
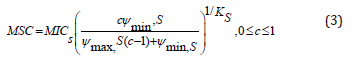
The population dynamics of the susceptible and resistant strains is captured in the following system of differential equations:
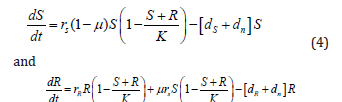
To include the change of antimicrobial concentrations over time (pharmacokinetics) into our model, we define the death rate to be dependent on the time-dependent antimicrobial concentration a(t):
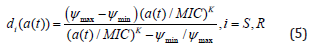
We assume a time-dependent pharmacokinetic function a(t) of the following form
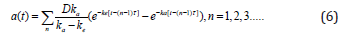
We define the treatment dose as the average concentration during the course of treatment:

Therefore, modeling hazard function can be written as
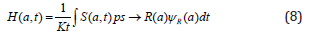
In order to generate predictions on antibiotics resistance based on pharmacodynamics, one of our main goals of the project, we made a number of simplifying assumptions. The pharmacodynamics are based on data of initial killing only. Moreover, we assume homogeneous populations over time and space. We implemented resistance, without considering whether resistance mutations are costly or mitigated by compensatory mutations. Our simulations suggest that resistance is limited in predicting resistance evolution based on pharmacodynamics. Expanding the framework to integrate tolerance and resistance is possible but would require pharmacodynamic estimates and additional functions. Another possible extension of our work would be to include pharmacodynamic estimates of resistant strains that change over time owing to compensatory mutations and to crossresistance or collateral sensitivity when exposed to combinations of antimicrobials. Finally, we assumed the same pharmacokinetics for all cases in our study. The future empirical work will inform realistic parameter estimates for pharmacokinetics. In all cases, however, the basis of any analysis concerning antibiotics resistance is the influence of individual pharmacodynamic parameters, for which we provide a framework.
Conclusion
Consequently, future studies should assess the value of even broader data for accurate prediction, ranging from transcriptome and proteome to other clinical and epidemiological data, such as cross-resistance and history of antibiotic therapy. Integrating these information sources from large isolate panels into a single predictive framework will lead to a rational basis for introducing machine learning in decision-making in public health.
Acknowledgement
I would like to appreciate great encouragement from Dr. Jae Hoon, Song who is a chairman of ANSORP as well as CHA Bio Group specially and this journal editor
Conflict of interest statement
Compliance with Ethical Standards The author reports no conflict of interest.
References
- Nesher L, Strahilevitz J (2019) Antibiotic stewardship in Israel-where are we headed in 2018. Harefuah 158(5): 321-326.
- Weinberger M (2019) Who's afraid of infectious diseases-a discipline with broad impact. Harefuah 158(5): 282-284.
- Holmes AH, Moore LS, Sundsfjord A, Steinbakk M, Regmi S, et al. (2016) Understanding the mechanisms and drivers of antimicrobial resistance. Lancet 387(10014): 176-187.
- Burnham CD, Leeds J, Nordmann P, Grady OJ, Patel J (2017) Diagnosing antimicrobial resistance. Nat Rev Microbiol 15(11): 697-703.
- McArthur AG, Waglechner N, Nizam F, Yan A, Azad MA, et al. (2013) The comprehensive antibiotic resistance database. Antimicrob Agents Chemother 57(7): 3348-3357.
- Zankari E, Hasman H, Cosentino S, Vestergaard M, Rasmussen S, et al. (2012) Identification of acquired antimicrobial resistance genes. J Antimicrob Chemother 67(11): 2640-2644.
- Bengtsson Palme J, Kristiansson E, Larsson DGJ (2018) Environmental factors influencing the development and spread of antibiotic resistance. FEMS Microbiol Rev 42(1): fux053.
- Manyi Loh C, Mamphweli S, Meyer E, Okoh A (2018) Antibiotic use in agriculture and its consequential resistance in environmental sources: Potential Public Health Implications. Molecules 23(4).
- Karkman A, Pärnänen K, Larsson DGJ (2019) Fecal pollution can explain antibiotic resistance gene abundances in anthropogenically impacted environments. Nat Commun 10(1): 80.
- Na Lu, Yongfei Hu, Liying Zhu, Xi Yang, Yeshi Yin, et al. (2014) DNA microarray analysis reveals that antibiotic resistance-gene diversity in human gut microbiota is age related. Sci Rep 4: 4302.
- Bengtsson-Palme J (2018) The diversity of uncharacterized antibiotic resistance genes can be predicted from known gene variants-but not always. Microbiome 6(1): 125.
- Pal C, Johan BP, Kristiansson E, Joakim L DG (2016) The structure and diversity of human, animal and environmental resistomes. Microbiome 4: 54.
- Ghosh TS, Gupta SS, Balakrish NG, Sharmila SM (2013) In Silico Analysis of antibiotic resistance genes in the gut microflora of individuals from diverse geographies and age-groups. PLoS One 8(12): e83823.
- Liu F, Zhu Y, Yi Y, Lu N, Zhu B, et al. (2014) Comparative genomic analysis of acinetobacter baumannii clinical isolates reveals extensive genomic variation and diverse antibiotic resistance determinants. BMC Genomics 15:1163.
- Peterson E, Kaur P (2018) Antibiotic resistance mechanisms in bacteria: relationships between resistance determinants of antibiotic producers, environmental bacteria, and clinical pathogens. Front Microbiol 9: 2928.
- McArthur AG, Waglechner N, Nizam F, Yan A, Azad MA, et al. (2013) The Comprehensive antibiotic resistance database. Antimicrob Agents Chemother 57(7): 3348-3357.
- Sommer MOA, Munck C, Toft Kehler RV, Andersson D (2017) Prediction of antibiotic resistance: Time for a new preclinical paradigm. Nat Rev Microbiol 15(11): 689-696.
- Biswas R, Panja AS, Bandopadhyay R (2019) Molecular mechanism of antibiotic resistance: The untouched area of future hope. Indian J Microbiol 59(2): 254-259.
- World Health Organization (2014) Antimicrobial resistance: Global report on surveillance 2014. WHO, Geneva, Switzerland.
- Cosgrove SE (2006) The relationship between antimicrobial resistance and patient outcomes: mortality, length of hospital stay, and health care costs. Clin Infect Dis 42(Suppl 2): S82-S89.
- Diaz Granados CA, Zimmer SM, Klein M, Jernigan JA (2005) Comparison of mortality associated with vancomycin-resistant and vancomycin-susceptible enterococcal bloodstream infections: A meta-analysis. Clin Infect Dis 41(3): 327-333.
- Sydnor ER, Perl TM (2011) Hospital epidemiology and infection control in acute-care settings. Clin Microbiol Rev 24(1): 141-173.
- Centers for Disease Control and Prevention (2013) Antibiotic resistance threats in the united states. Centers for Disease Control and Prevention, Atlanta, GA.
- The Review on Antimicrobial Resistance (2014) Antimicrobial resistance: tackling a crisis for the future health and wealth of nations.
- Clinical and Laboratory Standards Institute (2014) Performance standards for antimicrobial susceptibility testing; 24th informational supplement. CLSI document M100-S24. CLSI, Wayne, Pennsylvania, 34(1).
- Nannini EC, Singh KV, Arias CA, Murray BE (2013) In vivo effect of cefazolin, daptomycin, and nafcillin in experimental endocarditis with a methicillin-susceptible Staphylococcus aureus strain showing an inoculum effect against cefazolin. Antimicrob Agents Chemother 57(9): 4276-4281.
- Manson JM, Hancock LE, Gilmore MS (2010) Mechanism of chromosomal transfer of Enterococcus faecalis pathogenicity island, capsule, antimicrobial resistance, and other traits. Proc Natl Acad Sci USA 107(27): 12269-12274.
- Thomas CM, Nielsen KM (2015) Mechanisms of, and barriers to, horizontal gene transfer between bacteria. Nat Rev Microbiol 3(9): 711-721.
- D Costa VM, McGrann KM, Hughes DW, Wright GD (2006) Sampling the antibiotic resistome. Science 311(5759): 374-377.
- Bhullar K, Waglechner N, Pawlowski A, Koteva K, Banks ED, et al. (2012) Antibiotic resistance is prevalent in an isolated cave microbiome. PLoS One 7(4): e34953.
- Fajardo A, Martínez Martín N, Mercadillo M, Galán JC, Ghysels B, et al. (2008) The neglected intrinsic resistome of bacterial pathogens. PLoS ONE. 3(2): e1619.
- Martínez JL (2008) Antibiotics and antibiotic resistance genes in natural environments. Science 321(5887): 365-367.
- Dantas G, Sommer MO (2012) Context matters-the complex interplay between resistome genotypes and resistance phenotypes. Curr Opin Microbiol 15(5): 577-582.
- Fajardo A, Martínez JL (2008) Antibiotics as signals that trigger specific bacterial responses. Curr Opin Microbiol 11(2): 161-167.
- Dantas G, Sommer MO, Oluwasegun RD, Church GM (2008) Bacteria subsisting on antibiotics. Science 320(5872): 100-103.
- Toleman MA, Spencer J, Jones L, Walsh TR (2012) BlaNDM-1 is a chimera likely constructed in acinetobacter baumannii. Antimicrob. Agents Chemother 56(5): 2773-2776.
- Baquero F, Alvarez Ortega C, Martinez JL (2009) Ecology and evolution of antibiotic resistance. Environ Microbiol Rep 1(6): 469-476.
- Gullberg E, Cao S, Berg OG, Ilbäck C, Sandegren L, et al. (2011) Selection of resistant bacteria at very low antibiotic concentrations. PLoS Pathog 7(7): e1002158.
- Baquero F (2012) Metagenomic epidemiology: a public health need for the control of antimicrobial resistance. Clin Microbiol Infect 18(Suppl 4): 67-73.
- Cosgrove SE (2006) The relationship between antimicrobial resistance and patient outcomes: mortality, length of hospital stay, and health care costs. Clin Infect Dis 42(Suppl 2): S82-S89.
- Sydnor ER, Perl TM (2011) Hospital epidemiology and infection control in acute-care settings. Clin Microbiol Rev 24(1): 141-173.
- DiazGranados CA, Zimmer SM, Klein M, Jernigan JA (2005) Comparison of mortality associated with vancomycin-resistant and vancomycin-susceptible enterococcal bloodstream infections: a meta-analysis. Clin Infect Dis 41(3): 327-333.
- Nannini EC, Singh KV, Arias CA, Murray BE (2013) In vivo effect of cefazolin, daptomycin, and nafcillin in experimental endocarditis with a methicillin-susceptible Staphylococcus aureus strain showing an inoculum effect against cefazolin. Antimicrob Agents Chemother 57(9): 4276-4281.
- Manson JM, Hancock LE, Gilmore MS (2010) Mechanism of chromosomal transfer of enterococcus faecalis pathogenicity island, capsule, antimicrobial resistance, and other traits. Proc Natl Acad Sci USA 107(27): 12269-12274.
- Thomas CM, Nielsen KM (2005) Mechanisms of, and barriers to, horizontal gene transfer between bacteria. Nat Rev Microbiol 3(9): 711-721.
- Leclercq R (2002) Mechanisms of resistance to macrolides and lincosamides: nature of the resistance elements and their clinical implications. Clin Infect Dis 34: 482-492.
- Roberts MC (2008) Update on macrolide-lincosamidestreptogramin, ketolide, and oxazolidinone resistance genes. FEMS Microbiol Lett 282: 147-159.
- Katz L, Ashley GW (2005) Translation and protein synthesis: macrolides. Chem Rev 105(2): 499-528.
- Toh SM, Xiong L, Arias CA, Villegas MV, Lolans K et al. (2007) Acquisition of a natural resistance gene renders a clinical strain of methicillin-resistant Staphylococcus aureus resistant to the synthetic antibiotic linezolid. Mol Microbiol 64(6): 1506-1514.
- Locke JB, Zurenko GE, Shaw KJ, Bartizal K (2014) Tedizolid for the management of human infections: in vitro Clin Infect Dis 58(Suppl 1): S35-S42.
- Wilson DN (2014) Ribosome-targeting antibiotics and mechanisms of bacterial resistance. Nat Rev Microbiol 12(1): 35-48.
- World Health Organization (2014) Companion Handbook to the WHO Guidelines for the Programmatic Management of Drug-Resistant Tuberculosis. Geneva, Switzerland.
- Coll F, Phelan J, Hill-Cawthorne GA, Nair MB, Mallard K, et al. (2018) Genome-wide analysis of multi- and extensively drug-resistant mycobacterium tuberculosis. Nat Genet 50(2): 307-316.
- Alland D (2014) Pre-clinical development of an advanced genexpert test for drug resistant MTB. National Institute of Health, Newark, NJ, USA.
- Chakravorty S, Simmons AM, Rowneki M, Parmar H, Cao Y et al. (2017) The new Xpert MTB/RIF ultra: improving detection of mycobacterium tuberculosis and resistance to rifampin in an assay suitable for point-of-care testing. MBio 8(4): e00812- e00817.
- Hain Life Science (2018) Geno type MTBDR plus VER 2.0 -your test system for a fast and reliable way to detect MDR-TB pp. 1-14.
- Boehme CC, Nabeta P, Hillemann D, Nicol MP, Shenai S, et al. (2010) Rapid molecular detection of tuberculosis and rifampin resistance. N Engl J Med 363(11): 1005-1015.
- Laxminarayan R, Sridhar D, Blaser M, Wang M, Woolhouse M (2016) Achieving global targets for antimicrobial resistance. Science 353(6302): 874-875.
- McClure NS, Day T (2014) A theoretical examination of the relative importance of evolution management and drug development for managing resistance. Proc Biol Sci 281: 1797.
- World Health Organization (2014) The evolving threat of antimicrobial resistance: options for action. Geneva, Switzerland.
- Czaplewski L, Bax R, Clokie M, Dawson M, Fairhead H, et al. (2016) Alternatives to antibiotics: A pipeline portfolio review. Lancet Infect Dis 16(2): 239-251.
- Zasloff M (2002) Antimicrobial peptides of multicellular organisms. Nature 415(6870): 389-395.
- Hancock REW, Sahl HG (2006) Antimicrobial and host-defense peptides as new anti-infective therapeutic strategies. Nat Biotechnol 24(12): 1551-1557.
- Jochumsen N, Marvig RL, Damkiær S, Jensen RL, Paulander W, et al. (2016) The evolution of antimicrobial peptide resistance in Pseudomonas aeruginosa is shaped by strong epistatic interactions. Nat Commun 7: 13002.
- Fjell CD, Hiss JA, Hancock REW, Schneider G (2012) Designing antimicrobial peptides: form follows function. Nat Rev Drug Discov 11(1): 37-51.
- Ling LL, Schneider T, Peoples AJ, Spoering AL, Engels I et al. (2015) A new antibiotic kills pathogens without detectable resistance. Nature 517(7535): 455-459.
- Mohamed AF, Cars O, Friberg LE (2014) A pharmacokinetic/pharmacodynamic model developed for the effect of colistin on Pseudomonas aeruginosa in vitro with evaluation of population pharmacokinetic variability on simulated bacterial killing. J Antimicrob Chemother 69(5): 1350-1361.
- Firsov AA, Strukova EN, Shlykova DS, Portnoy YA, Kozyreva VK, et al. (2013) Bacterial resistance studies using in vitro dynamic models: The predictive power of the mutant prevention and minimum inhibitory antibiotic concentrations. Antimicrob Agents Chemother 57(10): 4956-4962.
- Drlica K, Zhao X (2007) Mutant selection window hypothesis updated. Clin Infect Dis 44(5): 681-688.
- Gullberg E, Cao S, Berg OG, Ilbäck C, Sandegren L, et al. (2011) Selection of resistant bacteria at very low antibiotic concentrations. PLoS Pathog 7(7): e1002158.
- Rodríguez-Rojas A, Makarova O, Rolff J (2014) Antimicrobials, stress and mutagenesis. PLoS Pathog 10(10): e1004445.
- Regoes RR, Wiuff C, Zappala RM, Garner KN, Baquero F, et al. (2004) Pharmacodynamic functions: A multiparameter approach to the design of antibiotic treatment regimens. Antimicrob Agents Chemother 48(10): 3670-3676.
- Cui J, Liu Y, Wang R, Tong W, Drlica K, et al. (2006) The mutant selection window in rabbits infected with staphylococcus aureus. J Infect Dis 194(11): 1601-1608.
- Fantner GE, Barbero RJ, Gray DS, Belcher AM (2010) Kinetics of antimicrobial peptide activity measured on individual bacterial cells using high-speed atomic force microscopy. Nat Nanotechnol 5(4): 280-285.
- Yu G, Baeder DY, Regoes RR, Rolff J (2017) Predicting drug resistance evolution: insights from antimicrobial peptides and antibiotics. Biol Sci 10: 1101.
- Greenfield BK, Shaked S, Marrs CF, Nelson P, Raxter I, et al. (2018) Modeling the emergence of antibiotic resistance in the environment: An analytical solution for the minimum selection concentration. Antimicrob Agents Chemother 62(3): e01686-016817.
- Mouton JW, Dudley MN, Cars O, Derendorf H, Drusano GL (2005) Standardization of pharmacokinetic/pharmacodynamic (PK/PD) terminology for anti-infective drugs: An update. J Antimicrob Chemother 55(5): 601-607.
- Andersson DI, Hughes D, Kubicek-Sutherland JZ (2016) Mechanisms and consequences of bacterial resistance to antimicrobial peptides. Drug Resist Update 26: 43-57.
- Day T, Read AF (2016) Does high-dose antimicrobial chemotherapy prevent the evolution of resistance? PLoS Comput Biol 12(1): e1004689.
- Krch MP (2008) Gillespie SSA: implementing the stochastic simulation algorithm in R. J Stat Softw 25(12): 1-18.
- R Core Team (2015) R: A language and for statistical computing: R Foundation for Statistical Computing, Vienna, Austria.
© 2019 Hyunjo Kim. This is an open access article distributed under the terms of the Creative Commons Attribution License , which permits unrestricted use, distribution, and build upon your work non-commercially.
Editor In Chief







.jpg)

























Signup for Newsletter

Quick Links
 Editorial Board Registrations
Editorial Board Registrations Submit your Article
Submit your Article Refer a Friend
Refer a Friend Advertise With Us
Advertise With UsOur Recent Edition

.jpg)



Top Editors




.jpg)

















.bmp)
.jpg)
.png)
.jpg)













.jpg)






.png)

.png)




.png)




Financial Support

Sponsors

Latest e-Books

Latest Video

 a Creative Commons Attribution 4.0 International License. Based on a work at www.crimsonpublishers.com.
Best viewed in
a Creative Commons Attribution 4.0 International License. Based on a work at www.crimsonpublishers.com.
Best viewed in