
- About Us
- Information
-

The Author ensures that the research has been conducted responsibly and ethically with adherence to all relevant regulations. read more..
- For Authors
- For Reviewer
- Manuscript Guidelines
- Membership
- Publication Ethics
-
- Journals
- Reprints
- e-Books
- Videos
- Policies
- Contact Us
 COVID-19
COVID-19
- Submissions



Full Text
Aspects in Mining & Mineral Science
Thermodynamics of Vacancy Formation in the CoCrFeMnNi High Entropy Alloy from DFT Calculations
Vsevolod I Razumovskiy1*, Daniel Scheiber1, Oleg Peil1, Andreas Stark2, Michael Mayer1 and Gerald Ressel1
1Materials Center Leoben Forschung GmbH, Roseggerstraße 12, Austria
2Helmholtz-Zentrum Hereon, Institute of Materials Physics, Max-Planck-Straße 1, Germany
*Corresponding author: Vsevolod I Razumovskiy, Materials Center Leoben Forschung GmbH, Roseggerstraße 12, Leoben, 8700, Austria
Submission: February 16, 2022;Published: March 09, 2022

ISSN 2578-0255Volume8 Issue5
Abstract
Vacancy formation in High Entropy Alloys (HEA) is an important aspect of their thermodynamic and kinetic theoretical description. Recent studies have shown that this topic can be approached from the side of density functional theory investigations for some concentrated alloys. However, DFT modeling of vacancy formation in HEA remains a challenging and often too complex task due to large sets of atomic configurations near a point defect that need to be accounted for in the calculations. Another challenge is related to an additional degree of complexity related to the paramagnetic state of some HEA such as the classical equimolar fcc CoCrFeMnNi. In this study, we demonstrate a way to reduce the computational complexity of point defect calculations in HEA and investigate the formation of thermal vacancies in the paramagnetic fcc CoCrFeMnNi alloy using DFT-based models and experimental X- ray diffraction analysis of the alloy crystal structure. We evaluate the effect of the paramagnetic state with respect to standard non-magnetic calculations. The calculated results predict a sizable contribution from the magnetic state with yet minor variations of the effective vacancy formation energy in the range from 1.66 at the room temperature up to 1.76eV at 1373K.
Keywords: High entropy alloys; Point defects; Density functional theory; Paramagnetism; Disordered alloys
Introduction
High Entropy Alloys (HEA) remain in focus of intensive research over the last decade due to their outstanding properties [1]. HEA are often defined as single phase alloys consisting of multiple base elements varying their composition in the range from 5 to 35 at. % [1]. The most known representative of HEA is the equiatomic face centered cubic CoCrFeMnNi - or Cantor - alloy [2]. The name originates from the first idea that the high mixing entropy stabilizes the single-phase solid solution [1,2]. However, nowadays, also multiphase alloys - mostly denoted as Complex Concentrated Alloys (CCA’s) - with varying elemental content exhibiting various atomic ordering effects are being considered as HEA as well. As they are envisioned to play an important role in the future of this alloy class [3,4] they are also studied in detail by computational methods [5]. Most of the kinetic processes in HEA are related to vacancy mediated diffusion, which is known to be retarded in some HEA and often referred to as the sluggish diffusion [1]. However, other studies report contradictory results, showing no significant change of the diffusion coefficient compared to the elemental metals [4], calling for detailed investigations of this phenomenon. Vacancy formation and migration processes have also been reported to be an important aspect in development of advanced irradiation resistant alloys for applications in nuclear fusion and -fission reactors [6]. During irradiation, vacancies are formed and actively participate in the main damage mechanisms in these materials [7,8]. Another application area of HEA that draws attention with regard to point defect formation is the production of oxide dispersion strengthened materials for applications at elevated temperatures such as in energy conversion or propulsion systems [9-11]. During mechanical alloying, the process of incorporating the oxide particles, vacancies seem to foster the dissolution or refinement of oxides leading to enhanced mechanical properties and shorter milling times and therefore a detailed knowledge of vacancy formation processes in these materials is of high interest [10-13].
Investigation of the vacancy formation in compositionally complex HEA using predictive density functional theory methods has drawn much attention in the recent years [14-24]. Compared to point defect formation in one-component materials, the problem of point defect formation in HEA is related to its dependence on the local chemical environment, which makes both experimental and theoretical investigations a challenging task. In the case of theoretical studies, vacancy formation in HEA is strongly connected to sampling of the atomic calculations over a set of crystal structures with various local chemical environments. This task alone represents a challenge for modern predictive density functional theory methods at 0K [25] and the most recent studies suggest employing machine learning approaches to provide a computationally efficient solution to the problem [14,24]. This problem gets even more complicated for magnetic systems, where the chemical disorder is often intertwined with the magnetic disorder that adds an extra degree of complexity to the theoretical description of the material that cannot be routinely modeled by most of the existing DFT codes. Adding the temperature dependence to this task, including both lattice expansion and anharmonic vibrations, that have been shown to be an important aspect of a theoretical evaluation of the vacancy formation, when comparing to experimental data [26-31], seems to be a prohibitively expensive task for most of the HEA systems from the computational point of view and therefore it has not been directly addressed so far to the best of our knowledge. Whereas an efficient general solution to the complete temperature/ composition/magnetic state ab initio description of HEA may still require some time to be implemented, a theoretical description of the vacancy formation in paramagnetic HEA can be approached from the side of existing mean-field approaches for the electronic structure and configurational thermodynamics calculations [32]. In Ref. [32], it has been shown that some simple mean-field considerations along with statistical methods can be efficiently used to describe the configurational contribution to the formation energy of vacancies in binary solid solutions at elevated temperatures. A special role in this investigation has been given to the effective vacancy formation energy, which was shown to be significantly lower than the mean value of the local vacancy formation energy calculated in most of DFT investigations and strongly dependent on temperature and configurational effects. In this work, we would like to extend and apply some parts of the aforementioned approach to a multicomponent system and demonstrate an example of the vacancy formation energy calculation in paramagnetic fcc CoCrFeMnNi HEA.
Methods
DFT computational details
The Nonmagnetic (NM) total energy calculations have been performed by the Projector Augmented Wave (PAW) method [33] implemented in the Vienna Ab Initio Simulation Package (VASP) [34,35]. The Perdew-Burke-Ernzerhof (PBE) form of the Generalized Gradient Approximation (GGA) [36] was used for the exchange-correlation energy. The energy cutoff was 270eV. The Brillouin zone integration was done using using the k-point mesh approximately equivalent to the 12×12×12 grid [37] for the 4-atom conventional fcc cubic unit cell. The magnetic contribution has been obtained by the EMTO (exact muffin-tin orbitals) [38-40]-LSGF [41] (locally self-consistent Green function method) method [42] which allows disordered local moment (DLM) supercell calculations within the Coherent Potential Approximation (CPA) [43,44]. All the self-consistent EMTO-LSGF calculations were performed by using an orbital momentum cutoff of lmax=3 for the partial waves in the spdf basis with the local interaction zone for electronic multiple scattering processes set to three nearest neighbor shells in the supercell calculations. The total energies were obtained using the GGA-PBE functional [36]. The magnetic entropy contribution at elevated temperatures has been included in a model form of longitudinal spin fluctuations (LSF) [45].
Vacancy formation energy
In contrast to one-element-based systems, vacancy formation in alloys depends on the local atomic environment around a lattice site where it is formed. In a general case, one can speak of the local vacancy formation energy distribution function g(Ef) that can be defined as
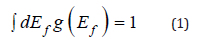
Here, the formation energy of each thermal vacancy, Ef, is calculated using two types of supercells designed to preserve the chemical composition of the alloy. Here, we have used a 4×4×4 supercell made by replication of the 4-atom conventional fcc cell (256 sites) to model a vacancy in the 5-component equimolar CrMnFeCoNi alloy with 20% (51 atom) of each atomic species. Another 5×5×5 supercell made by replication of the primitive 1-atom fcc cell (125 sites) has been used to model the bulk reference with ideal lattice (no vacancy). In both supercells, the atoms have been distributed on the lattice to make sure that the pair correlation functions between all atomic species on the first two coordination spheres are as close as possible to the values of a disordered alloy. Due to cell-size limitations, it was impossible to reach identical short range order values for both cell types and the SRO values of the smaller (125 site) cell have been adopted in both calculations. In this case, the vacancy formation energy of an ith alloy configuration reads:
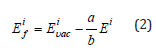
where a is the number of atoms in a cell with a vacancy (255) and b is the number of atoms in a cell without the vacancy (125). The convergence of this configuration with respect to the SRO has been checked by a test calculation of a 5×5×5 supercell made by replication of the 4-atom conventional fcc cell (500 sites) with the SRO parameters of the 256-site cell. The vacancy formation energy difference between two supercell setups was less than 0.008eV. According to Ref. [32], the local vacancy formation energy distribution function g(E) in an equiatomic binary alloy can be approximated by the discreet binomial distribution. In the case of an equiatomic multicomponent alloy, this distribution has to be replaced by a multinomial distribution function:
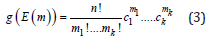
where k is the number of alloy components; ck is the atomic
fraction of the kth component (Σck = 1) ; n is the number of nearest
neighbors in the considered coordination shell (12 in the case of the
1st nearest neighbor shell in the fcc crystal structure); each m=(m1,·
· ·,mk) is the number of type k atoms in the given coordination shell
(their sum equals to n); E(m)=(E1,….,Ek) are the vacancy formation
energies as a function of m calculated using Eq. 2, which can also be
expressed as 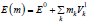 where E0 is the lowest local vacancy
formation energy corresponding to n=0 and Vk
1 is the interaction
energy between the vacancy and kth alloy component in the 1st
coordination shell [32]. The latter can be determined either by the
slope of the E(mk), assuming that interatomic interaction in the
system is relatively weak, or by the multivariate regression analysis
of E(m). En equivalent representation of the vacancy formation
energy distribution function from (32) is:
where E0 is the lowest local vacancy
formation energy corresponding to n=0 and Vk
1 is the interaction
energy between the vacancy and kth alloy component in the 1st
coordination shell [32]. The latter can be determined either by the
slope of the E(mk), assuming that interatomic interaction in the
system is relatively weak, or by the multivariate regression analysis
of E(m). En equivalent representation of the vacancy formation
energy distribution function from (32) is:
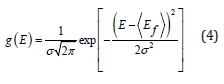
where σ is its standard deviation, Ef is the mean value of the vacancy formation energy. This expression can be adopted to obtain the effective local vacancy formation energy as:
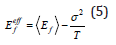
Experimental details
Investigated powder material was the equiatomic face centered cubic CoCrFeMnNi alloy exhibiting a mean particle size of 400μm. It was a powder milled for 48h under high vacuum in a specifically constructed attritor mill. In situ X-ray diffraction patterns of the equiatomic face centered cubic CoCr-FeMnNi alloy were measured at the HEMS beamline of PETRA III [46] at Deutsches Elektronen- Synchrotron (DESY) in Hamburg using high-energy synchrotron radiation. The powder was put in a Nb-capsule and measured at 25 °C as well as at 1100 °C with a heating rate of 100K/min in-between. For synchrotron measurements a detector distance of 1.4m and a wavelength of 0.14235Å was used. For evaluation regarding lattice parameter the patterns were fitted using a Pseudo-Voigt model and the peak positions were measured from the maximum of the fit. Due to planar faults present in the investigated material after milling, asymmetric peak shifts occurred and were corrected using the theoretical formula described by Warren [47] and an error minimization approach for all measured peaks. By this, the effect of planar faults on the lattice parameter was corrected.
Results and Discussion
DFT calculations of the vacancy formation in multicomponent alloys is not a trivial task that may become unaffordable expensive as the number of alloy components increases. Currently available technical solutions to this problem usually involve either supercell techniques that rely on averaging over large-enough sets of atomic configurations or on CPA-based methods based on the mean-filed representation of an alloy. Both methods have their advantages and disadvantages when it comes to the calculation of defects in an alloy. In this paper, we demonstrate a computational approach for DFT calculations of the vacancy formation energy that can combine both methods and benefit from their strong sides. Our computational procedure consists of two main steps: i) calculation of the minimal set of supercells representing the equimolar disordered alloy with a vacancy in the none-magnetic (NM) state; ii) calculation of the magnetic contribution to the vacancy formation energy using DLMCPA. Here, we focus on formation of thermal vacancies which are formed without changing the chemical composition of the alloy. Therefore, we have used two different supercells to represent the system with and without a vacancy chosen in a way that they both have the same equimolar chemical composition (of 20 at. % for each atomic species) as described in Section 2.2. In (Figures 1 & 2), we show the local vacancy formation energy distribution function and the formation energy dependence on the number of nearest neighbor atoms of each individual alloy component, respectively. These results have been obtained in the NM calculations using the Room Temperature (RT) lattice parameters obtained from the XRD measurements (Table 1). Figure 1 also shows the scatter of the vacancy formation energies that covers the range between 2 and 3eV.
Table 1: Alloy lattice parameters (a in Å), vacancy formation energies (in eV) with corresponding temperatures (in K) and the standard deviations of the local vacancy formation energies using Eq. 3 fit to DFT data. The correction from paramagnetic state from LSF and DLM is denoted by Δ(Ef).

Figure 1: Local vacancy formation energy distribution function g(E) obtained from DFT calculations (orange) and approximated by Eq. 4 (blue).

Figure 2: Local vacancy formation energy dependence on a number of nearest neighbor atoms of alloy components in the supercell. Linearly extrapolated data are shown with solid lines. Symbol and line coloring in the graphs corresponds to the atom coloring in a schematic representation of the supercell used in the calculations.

A more detailed analysis of the distribution of the local vacancy formation energies with respect to the number of nearest neighbor atoms of different sorts can be seen in Figure 2. Here, by analogy with binary alloys [32], we analyze individual contributions of each alloy component to the vacancy formation energy in the alloy. The slopes of the linear fits to these data correspond to the interaction energy of a vacancy with the atoms of each selected alloying elements, assuming that interactions between the atoms themselves are relatively weak. The number on the x-axis in Figure 2 corresponds to the number of atoms in the first coordination shell varying from 1 to 12. There has been no site surrounded by 12 nearest neighbor atoms of the same species in the analyzed supercells and we have used a set of additional calculations where the cell volume and fcc structure were kept fixed, but all atoms were replaced solely by one type of atomic species to mimic the limiting case of 12 nearest neighbor atoms of the same sort. The results of this investigation suggest that an increase of the number of Cr, Ni and Co atoms at the first coordination shell around the vacancy reduces the Ef on average, whereas Mn and Fe slightly increase it. This result can also be interpreted in a way that the vacancy is likely to be surrounded by the atoms of these sorts in the alloy. Mn has virtually no effect on the Ef and the strongest effect belongs to Ni that can lower the Ef by up to 20% in our estimate of the limiting case of its 100% occupation of the 1st coordination shell and only by around 10% at the 20% concentration corresponding to its content in the Cantor alloy. These results also indicate that the interatomic interactions in the considered alloy are relatively small compared to the formation energy of a vacancy itself. The results on the vacancy interaction energies with alloying elements allow us to reconstruct the normal multinomial distribution function using Eq. 3. Assuming that shape of this distribution function is generic for concentrated equimolar alloys [32], one can use it to obtain an accurate (physical) fit of the calculated in DFT data and extract necessary for calculating Ef eff standard deviation and mean formation energy values (Table 1). The result of the fit is shown in (Figure 1) in blue and indicates some differences between the normal and DFT-based distributions, which are most likely related to the limited set of selected atomic configurations for DFT calculations. However, we observe a rather fast convergence of the results of the fit even if one uses a very limited set of DFT data: Already a set of 11 atomic configurations selected according to Sec. 2.2 is sufficient to get the mean formation energy value within 0.012eV and the σ within 0.019 error and the results for ¿40 alloy configurations calculated in DFT lead to practically no change in the fit parameters. This way, we see a great advantage in the use of Eq. 3 for analysis of the vacancy formation in equimolar alloys as it can substantially reduce the computational cost related to the DFT calculations of various defect configurations.
Figure 3: Effective vacancy formation energy as a function of temperature, calculated at the RT and HTL DFT input data (solid lines). Vertical dashed lines represent the experimental from Ref. (22) (blue) and the mean 0K DFT values (black). Two circles connected with a red dashed line represent the results of the effective vacancy formation energy predictions based on both DFT and model calculations done at the corresponding temperatures (298 and 1373K) suitable for comparison to the experimental data.

The calculated values so far have been obtained in the nonmagnetic state approximation. However, the alloy under consideration is paramagnetic and therefore we have applied the DLM and LSF models to calculate the contribution of paramagnetism to the calculated vacancy formation energies in the NM state. The calculations have been done at two representative temperatures denoted as RT (room temperature, 298K) and HT (high temperature, 1373K) at the corresponding lattice parameters (RTL and HTL respectively) measured in XRD (Table 1). The results show that the NM calculations overestimate the Ef by 0.32 and 0.53eV (Table 1) compared to the DLM RTL+LSF and HTL+LSF calculations. We have applied this correction to our NM calculations to calculate the effective local vacancy formation energy Ef eff using Equation 5 and to compare it to the experiment. An important point in this analysis is that the effective local vacancy formation energy is temperature dependent and that it is always (at T<∞) smaller than the mean vacancy formation energy value, ⟨Ef ⟩ (see Equation 5). These results can be clearly seen in (Figure 3), where Ef eff is plotted along with the mean ⟨Ef ⟩ and experimental vacancy formation energy values, as well as in Table 1. The Ef eff values presented in the table for RT and HTL calculations yield 1.66 and 1.76eV respectively. These results are very close to the experimental value of 1.69eV [22], which refers to the positron lifetime measurements. In Ref. [22], the authors were able to single out the formation of thermal vacancies that were found to be dominant in the CoCrFeMnNi HEA in the temperature interval from 1181 to 1423K and declared that the effective formation energy of them remain virtually a constant within this interval. If we connect two calculated points referring to RT and HTL in (Figure 3), we see that this conclusion is consistent with the theoretical prediction of this work, where we can see only a minor change in Ef eff as a function of temperature. Here, we must note that Equation 5 is strictly speaking valid in the high temperature regime, where the Ef eff changes are indeed linear. In the limit of T→0, the strong deviations from linearity are related to its analytical form, which leads to most likely artificial reduction of Ef eff and therefore underestimation of the Ef eff at RT in our calculations. Another important aspect is the partially neglected effect of the atomic vibrations that may have an impact on the resulting Ef eff values at high temperatures [28-31]. These effects in the selected alloy need to be investigated further in more detail, though they may have their main contribution to the vacancy formation in the considered system through the lattice expansion that is included in the calculation via adaptation of the experimental lattice constants. The remaining (not included in this work) part of it may have a minor impact on the thermal vacancy formation in the considered alloy, as we can see from the comparison of our results to the experimental data.
Conclusion
Vacancy formation in multicomponent paramagnetic alloys is a complex topic for density functional theory investigations. However, the complexity of the problem can be reduced by the use of a number of model steps that can make such calculations a feasible task. In this paper, we demonstrate the applicability of existing theoretical approaches for DFT-based calculations of the effective vacancy formation energy calculations and extend them to the paramagnetic fcc CoCrFeMnNi HEA. We perform two sets of DFT-based calculations corresponding to the room and high temperature material states and analyze the obtained vacancy formation energies as a function of thermal lattice expansion and the magnetic state. We show that the account of the magnetic degrees of freedom in the calculations leads to a sizable contribution to the formation energy of the defects. Our results show that the formation energy of thermal vacancies in the paramagnetic fcc CoCrFeMnNi HEA varies little as a function of temperature taking values from 1.66 to 1.76eV between 298 and 1373K, which agrees well with available experimental data.
Acknowledgment
The authors gratefully acknowledge the financial support under the scope of the COMET program within the K2 Center “Integrated Computational Material, Process and Product Engineering (ICMPPE)” (Project No 859480). This program is sup- ported by the Austrian Federal Ministries for Climate Action, Environment, Energy, Mobility, Innovation and Technology (BMK) and for Digital and Economic Affairs (BMDW), represented by the Austrian research funding association (FFG), and the federal states of Styria, Upper Austria and Tyrol. VIR and OP are grateful to Prof. Andrei Ruban for fruitful discussions of the methodology he published in Ref. [32]. The computational results have been achieved using the Vienna Scientific Cluster (VSC). This work has been carried out within the framework of the EURO fusion consortium and has received funding from the Euratom research and training programme 2014-2018 and 2019-2020 under grant agreement No 633053. The views and opinions expressed herein do not necessarily reflect those of the European Commission.
References
- Tsai MH, Yeh JW (2014) High-entropy alloys: A critical review. Materials Research Letters 2(3): 107-123.
- Cantor B, Chang I, Knight P, Vincent A (2004) Microstructural development in equiatomic multicomponent alloys. Materials Science and Engineering: A 375-377: 213-218.
- Murty BS, Yeh JW, Ranganathan S, Bhattacharjee P (2019) High-entropy alloys. Elsevier, Netherlands.
- Miracle DB, Senkov ON (2017) A critical review of high entropy alloys and related concepts. Acta Materialia 122: 448-511.
- Biermair F, Razumovskiy VI, Ressel G (2022) Influence of alloying on thermodynamic properties of AlCoCrFeNiTi high entropy alloys from DFT calculations. Computational Materials Science 202: 110952.
- Mansur L (1994) Theory and experimental background on dimensional changes in irradiated alloys. Journal of Nuclear Materials 216: 97-123.
- Stoller R, Odette G, Wirth B (1997) Primary damage formation in bcc iron. Journal of Nuclear Materials 251: 49-60.
- Nordlund K, Ghaly M, Averback RS, Caturla M, de la Rubia DT, et al. (1998) Defect production in collision cascades in elemental semiconductors and fcc metals. Phys Rev B 57(13): 7556-7570.
- Ressel G, Parz P, Fian A, Holec D, Primig S, et al. (2014) On the behavior of yttria/yttrium during mechanical alloying of a Fe-Y2O3 model alloy system. Advanced Materials Research 922: 598-603.
- Ressel G, Holec D, Fian A, Mendez-Martin F, Leitner H (2014) Atomistic insights into milling mechanisms in an Fe–Y2O3 model alloy. Applied Physics A 115(3): 851-858.
- Alinger M, Glade S, Wirth B, Odette G, Toyama T, et al. (2009) Positron annihilation characterization of nanostructured ferritic alloys. Materials Science and Engineering: A 518(1): 150-157.
- Ressel G (2014) New findings on the mechanisms during mechanical alloying of a Fe-Y2O3 model alloy and an oxide dispersion strengthened high-alloyed steel.
- Mayer M, Ressel G, Svoboda J (2021) The effect of cryogenic mechanical alloying and milling duration on powder particles’ microstructure of an oxide dispersion strengthened FeCrMnNiCo high-entropy alloy, Metallurgical and Materials Transactions A pp. 1-12.
- Manzoor A, Arora G, Jerome B, Linton N, Norman B, et al. (2021) Machine learning based methodology to predict point defect energies in multi-principal element alloys. Frontiers in Materials 8: 129.
- Manzoor A, Pandey S, Chakraborty D, Phillpot SR, Aidhy DS (2018) Entropy contributions to phase stability in binary random solid solutions. npj Computational Materials 4(1): 1-10.
- Ikeda Y, Grabowski B, Kormann F (2019) Ab initio phase stabilities and mechanical properties of multicomponent alloys: A comprehensive review for high entropy alloys and compositionally complex alloys. Materials Characterization 147: 464-511.
- Middleburgh S, King D, Lumpkin G, Cortie M, Edwards L (2014) Segregation and migration of species in the CrCoFeNi high entropy alloy. Journal of Alloys and Compounds 599: 179-182.
- Chen W, Ding X, Feng Y, Liu X, Liu K, et al. (2018) Vacancy formation enthalpies of high-entropy FeCoCrNi alloy via first-principles calculations and possible implications to its superior radiation tolerance. Journal of Materials Science & Technology 34(2): 355-364.
- Zhao S, Egami T, Stocks GM, Zhang Y (2018) Effect of d electrons on defect properties in equiatomic NiCoCr and NiCoFeCr concentrated solid solution alloys. Physical Review Materials 2(1): 013602.
- Velisa G, Wendler E, Zhao S, Jin K, Bei H, et al. (2018) Delayed damage accumulation by a thermal suppression of defect production in concentrated solid solution alloys. Materials Research Letters 6(2): 136-141.
- Xu Q, Guan H, Zhong Z, Huang S, Zhao J (2021) Irradiation resistance mechanism of the CoCrFeMnNi equiatomic high-entropy alloy. Scientific Reports 11(1): 608.
- Sugita K, Matsuoka N, Mizuno M, Araki H (2020) Vacancy formation enthalpy in CoCrFeMnNi high-entropy alloy. Scripta Materialia 176: 32-35.
- Vaidya M, Sen S, Zhang X, Frommeyer L, Rogal L, et al. (2020) Phenomenon of ultra-fast tracer diffusion of Co in HCP high entropy alloys. Acta Materialia 196: 220-230.
- Grabowski B, Ikeda Y, Srinivasan P, Kormann F, Freysoldt C, et al. (2019) Ab initio vibrational free energies including anharmonicity for multicom- ponent alloys. npj Computational Materials 5(1): 80.
- Aitken ZH, Sorkin V, Zhang YW (2019) Atomistic modeling of nanoscale plasticity in high-entropy alloys. Journal of Materials Research 34(9): 1509-1532.
- Gong Y, Grabowski B, Glensk A, Kormann F, Neugebauer J, et al. (2018) Temperature dependence of the Gibbs energy of vacancy formation of fcc Ni. Phys Rev B 97: 214106.
- Freysoldt C, Grabowski B, Hickel T, Neugebauer J, Kresse G, et al. (2014) First-principles calculations for point defects in solids. Reviews of Modern Physics 86(1): 253.
- Glensk A, Grabowski B, Hickel T, Neugebauer J (2014) Breakdown of the arrhenius law in describing vacancy formation energies: The importance of local anharmonicity revealed by ab initio thermodynamics. Phys Rev X 4: 011018.
- Gong Y, Grabowski B, Glensk A, Kormann F, Neugebauer J, et al. (2018) Temperature dependence of the gibbs energy of vacancy formation of fcc Ni. Physical Review B 97(21): 214106.
- Zhang X, Grabowski B, Hickel T, Neugebauer J (2018) Calculating free energies of point defects from ab initio. Computational Materials Science 148: 249-259.
- Ishibashi S, Ikeda Y, Kormann F, Grabowski B, Neugebauer J (2020) Correlation analysis of strongly fluctuating atomic volumes, charges, and stresses in body-centered cubic refractory high-entropy alloys. Physical Review Materials 4(2): 023608.
- Ruban AV (2016) Thermal vacancies in random alloys in the single-site mean-field approximation. Physical Review B 93(13): 134115.
- Blochl PE (1994) Projector augmented-wave method. Phys Rev B 50: 17953-17979.
- Kresse G, Hafner J (1993) Ab initio molecular dynamics for open-shell transition metals. Phys Rev B 48: 13115-13118.
- Kresse G, Furthmuller J (1996) Efficiency of ab-initio total energy calculations for metals and semiconductors using a plane-wave basis set. Comput Mater Sci 6(1): 15-50.
- Perdew JP, Burke K, Ernzerhof M (1996) Generalized gradient approximation made simple. Phys Rev Lett 77: 3865-3868.
- Monkhorst HJ, Pack JD (1976) Special points for brillouin-zone integrations. Phys Rev B 13: 5188-5192.
- Vitos L, Skriver H, Johansson B, Kollar J (2000) Application of the exact muffin-tin orbitals theory: the spherical cell approximation. Comput Mater Sci 18(1): 24- 38.
- Vitos L (2001) Total-energy method based on the exact muffin-tin orbitals theory. Phys Rev B 64: 014107.
- Vitos L, Abrikosov IA, Johansson B (2001) Anisotropic lattice distortions in random alloys from first-principles theory. Phys Rev Lett 87: 156401.
- Abrikosov IA, Niklasson AMN, Simak SI, Johansson B, Ruban AV, et al. (1996) Order-N green’s function technique for local environment effects in alloys. Phys Rev Lett 76: 4203-4206.
- Peil OE, Ruban AV, Johansson B (2012) Self-consistent supercell approach to alloys with local environment effects. Phys Rev B 85: 165140.
- Soven P (1967) Coherent-potential model of substitutional disordered alloys. Phys Rev 156: 809-813.
- Gyorffy BL (1972) Coherent-potential approximation for a nonoverlapping-muffin-tin- potential model of random substitutional alloys. Phys Rev B 5: 2382-2384.
- Ruban AV, Dehghani M (2016) Atomic configuration and properties of austenitic steels at finite temperature: Effect of longitudinal spin fluctuations. Phys Rev B 94: 104111.
- Schell N, King A, Beckmann F, Fischer T, Muller M, et al. (2014) The high energy materials science beamline (hems) at petra iii, In: Mechanical Stress Evaluation by Neutrons and Synchrotron Radiation VI, Vol. 772 of Materials Science Forum, Trans Tech Publications Ltd, pp. 57-61.
- Warren BE (1959) X-ray studies of deformed metals. Progress in Metal Physics 8: 147-202.
© 2022 Vsevolod I Razumovskiy. This is an open access article distributed under the terms of the Creative Commons Attribution License , which permits unrestricted use, distribution, and build upon your work non-commercially.
Editor In Chief







.jpg)

























Signup for Newsletter

Quick Links
 Editorial Board Registrations
Editorial Board Registrations Submit your Article
Submit your Article Refer a Friend
Refer a Friend Advertise With Us
Advertise With UsOur Recent Edition

.jpg)



Top Editors




.jpg)

















.bmp)
.jpg)
.png)
.jpg)













.jpg)






.png)

.png)




.png)




Financial Support

Sponsors

Latest e-Books

Latest Video

 a Creative Commons Attribution 4.0 International License. Based on a work at www.crimsonpublishers.com.
Best viewed in
a Creative Commons Attribution 4.0 International License. Based on a work at www.crimsonpublishers.com.
Best viewed in